
ABSTRACT
Previously we demonstrated that endometrium-derived human mesenchymal stem cells (hMESCs) via activation of the ATM/p53/p21/Rb pathway enter the premature senescence in response to oxidative stress. Down regulation effects of the key components of this signaling pathway, particularly ATM and p53, on a fate of stressed hMESCs have not yet been investigated. In the present study by using the specific inhibitors Ku55933 and Pifithrin-α, we confirmed implication of both ATM and p53 in H2O2-induced senescence of hMESCs. ATM or p53 down regulation was shown to modulate differently the cellular fate of H2O2-treated hMESCs. ATM inhibition allowed H2O2-stimulated hMESCs to escape the permanent cell cycle arrest due to loss of the functional ATM/p53/p21/Rb pathway, and induced bypass of mitosis and re-entry into S phase, resulting in tetraploid cells. On the contrary, suppression of the p53 transcriptional activity caused a pronounced cell death of H2O2-treated hMESCs via autophagy induction. The obtained data clearly demonstrate that down regulation of ATM or p53 shifts senescence of human endometrial stem cells toward tetraploidization or autophagy.
Introduction
Already six years past since the first evidence that human mesenchymal stem cells may undergo premature senescence in response to sublethal stress.Citation1 To date this phenomenon seems to be of a great importance due to the obvious fact that adult stem cell senescence is equal to the reduction of their regenerative ability, what directly concerns the effectiveness of their potential clinical application.Citation2,3,4,5 Cellular senescence is typically defined as a process in which cells cease dividing and undergo distinctive phenotypic alterations, including enlarged and flattened morphology, increased SA-β-Gal staining as well as the profound secretome changes termed senescence-associated secretory phenotype (SASP).Citation6,7 According to the recent data, senescent cells through autocrine/paracrine pathways may initiate premature senescence or even transformation of the neighboring cells,Citation8,9 what in context of adult stem cells is of a particular importance as it may limit their use in regenerative medicine. These notions raise a question of the elimination of aging cells from the population in order to prevent further senescence expansion.
It is well known that cell aging may be triggered either by telomere shorteningCitation10 or by the variety of stresses;Citation11,12 however despite the nature of the senescence inductor, the typical “starting point” is the DNA damage response (DDR) activation.Citation13,14 Although the initial goal of the DDR is to repair damaged DNA and restart the cell cycle, in case of irreparable damage it eventually induces an irreversible cell cycle arrest leading to senescence, or programmed cell death. DDR is a signaling pathway mediated by the phosphoinositide-3-kinase (PI-3K)-related protein kinases (PIK kinases) including ataxia-telangiectasia mutated (ATM), ATM and RAD3-related (ATR) and DNA-dependent protein kinase (DNA-PK). In undamaged cells, ATM is inactive however following DNA damage it immediately undergoes autophosphorylation, resulting in the formation of the active ATM monomers.Citation15,16,17 Once activated ATM is recruited to the sites of the DNA damage and initiates cell-cycle progression arrest through phosphorylation of direct downstream targets.
One of the most important ATM substrate is a tumor suppressor protein p53.Citation6 Following activation p53 is translocated into the nuclei where it modulates transcription of various genes. Due to the differential activation of target genes p53 governs pathways that direct cells either to cell cycle arrest, senescence, or apoptosis, thus preventing the propagation of damaged DNA.Citation18 Crucial transcriptional target and mediator of p53-dependent senescence is a cyclin-dependent kinase inhibitor – p21.Citation19 An enhanced expression of p21 leads to hypophosphorylation, and thus activation, of retinoblastoma protein (Rb) what in turn results in cell cycle and proliferation arrest.Citation20 Noteworthy, the described above DDR-mediated cell cycle arrest typically concerns to senescence initiation, however for further development toward irreversible, phenotypically complete senescence ATM/p53/p21/Rb pathway should be held in an active state long after senescence initiation.Citation21,22,23
Both ATM and p53, being critical regulators of cell fates after DNA damage, may induce a variety of cellular responses, including induction of cell cycle arrest, DNA repair, maintenance of genomic stability, induction of premature senescence and cell death.Citation24,25 Published data concerning cellular responses to ATM down regulation are rather controversial. In non transformed human senescent cells down regulation of ATM signaling most commonly leads to cell cycle re-entry and proliferation recovery,Citation26,27,28 whereas in both senescent tumor and hematopoietic progenitor cells it triggers apoptosis.Citation29,30.Citation31,32,33 The modern knowledge regarding the consequences of ATM inhibition in senescent human mesenchymal stem cells is very limited. In γ-irradiated human mesenchymal stem cells (MSC) isolated from dental pulp and periodontal ligament, early changes in DDR signaling induced by ATM activity suppression were evaluated.Citation34 The effects of p53 inhibition on the fate of senescent cells are rather diverse. Depending on the cell context, p53 inactivation has been shown to provoke either senescence reversal or autophagy.Citation35,36,37,38
In the recent study we have convincingly shown that human endometrium-derived mesenchymal stem cells (hMESCs) under oxidative stress enter the premature senescence that is accompanied by the activation of the main DDR members, including ATM, and by the irreversible growth arrest via the functional activation of the p53/p21/Rb pathway.Citation23 hMESCs are the perspective source of mesenchymal stem cells for transplantation in cell-based therapy.Citation39,40 However a loss of regenerative potential during senescence in oxidative stress conditions, as well as the senescence expansion due to SASP may dramatically diminish the effectiveness of transplantation. In this regard, down regulation of either ATM or p53, implicated in hMESCs senescence managing, may be considered as a strategy directed to eliminate potentially dangerous senescent cells. Thus, the present study aimed to elucidate the impact of ATM and p53 inhibition on the cell fate of the senescent hMESCs.
Results
ATM inhibition does not prevent H2O2-induced senescence of hMESCs
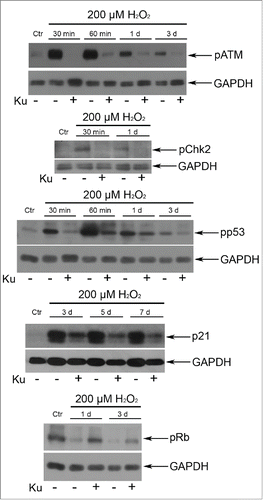
In order to elucidate a possible role of ATM kinase in the regulation of hMESCs senescence, we applied a specific inhibitor of ATM kinase activity – Ku55933 (Ku).Citation29 Cell pretreatment with 10 μM Ku had no significant effect on cell viability, but effectively blocked H2O2-induced ATM phosphorylation (). Taking into account our previous data that persuasively indicate the key role of ATM/Chk2/p53/p21/Rb pathway in the initiation and development of the hMESCs senescence,Citation23 first we investigated the effects of ATM blocking on function of this signaling cascade. As expected, ATM inhibition resulted in the phosphorylation decrease of direct ATM targets - Chk2 and p53. In Ku-treated cells we observed a prolonged down-regulation of p21 expression and slight increase in Rb phosphorylation compared with H2O2-treated hMESCs (). Thus, ATM activity suppression led to the functional inactivation of Chk2/p53/p21/Rb pathway that persisted for a long time after senescence initiation.
Figure 1. Dose-dependent effects of Ku application. (A) Cells were either treated or not with Ku at indicated concentrations. The percentage of viable cells was evaluated in 24 h after treatment using MTT assay as described in “Materials and Methods” section. Results are shown as a percent of control. M ± SD, N = 3. (B) Immunoblot analysis of H2O2-induced ATM phosphorylation. Representative results of the three experiments are shown in the figure. (Ctr – control). GAPDH was used as loading control.
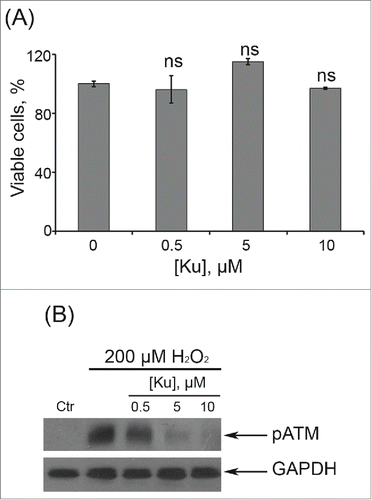
Figure 2. Application of 10 μM Ku led to the down regulation of the ATM/Chk2/p53/p21/pRb pathway in H2O2-treated hMESCs. The functional status of the tested proteins was detected by Western blotting with the use of specific antibodies. Representative results of the three experiments are shown in the figure. GAPDH was used as loading control.
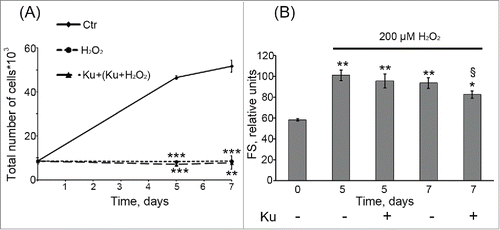
Based on these observations further we checked whether ATM inhibition may reverse the proliferation arrest induced by H2O2. Interestingly, pretreatment with Ku had no effect on cell proliferation compared with those exposed to H2O2 alone (). Moreover, blocking ATM activity had no influence on senescent phenotype of H2O2-treated cells, which was characterized by cell hypertrophy () and enhanced SA-β-Gal staining (data not shown). Thus, despite the down regulation of Chk2/p53/p21/Rb pathway, responsible for senescence initiation and development, ATM inhibition was not able to prevent H2O2-induced senescence of hMESCs.
Figure 3. Cell treatment with 10 μM Ku was not able to prevent H2O2-induced senescence of hMESCs. (A) Ku had no effect on the cell proliferation of H2O2-treated hMESCs. Cell number was determined by FACS at indicated time points (M ± SD, N = 3, **p<0.01, ***p<0.005, versus control). (B) ATM inhibition with Ku did not prevent H2O2-induced increase in the cell size. Forward scatter (FS) reflects the average cell size. M ± SD, N = 3, *p<0.05, **p<0.01, versus control, §p<0.05, versus H2O2-treated cells. Ctr – control. “Cell treatment with 10 μM Ku was not able to prevent H2O2-induced senescence of hMESCs. (A) Ku had no effect on the cell proliferation of H2O2-treated hMESCs. Cell number was determined by FACS at indicated time points (M ± SD, N = 3, **p<0.01, ***p<0.005, versus control). (B) ATM inhibition with Ku did not prevent H2O2-induced increase in the cell size. Forward scatter (FS) reflects the average cell size. M ± SD, N = 3, *p<0.05, **p<0.01, versus control, §p<0.05, versus H2O2-treated cells. Ctr – control.
Blocking ATM activity leads to the prolonged G2/M accumulation of senescent hMESCs
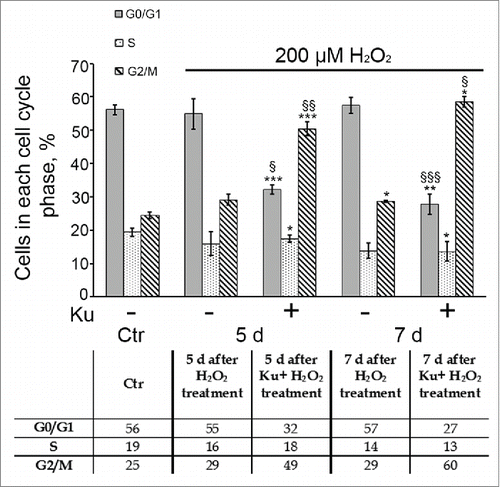
Earlier we have shown that hMESCs treatment with 200 μM H2O2 led to the prolonged irreversible cell cycle arrest in all phases.Citation41 Therefore further we checked whether cell pretreatment with Ku might affect H2O2-induced cell cycle arrest of hMESCs. Analysis of the cell cycle phase distribution revealed a persistent Ku-induced accumulation of H2O2-treated cells in G2/M phase. The increase of the cell proportion in the G2/M phase (from 50% at day 5 up to 60% at day 7) indicates the absence of a multiphase cell cycle block after Ku treatment (). In contrast, H2O2-treated cells with intact ATM displayed the expected cell cycle arrest in all phases, and besides the cell cycle phase ration in such cells remained constant at least for 7 d. It should be noted that in the absence of the oxidative stimulation, Ku had no effect on the cell cycle distribution of hMESCs during the investigated time frame ().
Figure 4. Down regulation of ATM activity by Ku resulted in the prolonged G2/M accumulation of H2O2-treated hMESCs. Flow cytometric analysis of cell cycle phase distribution: the percentage of cells in the G0/G1, S, and G2/M phases. M ± SD, N = 3, *p<0.05, ***p<0.005, versus control, §p<0.05, §§p<0.01, §§§p<0.005, versus H2O2-treated cells. Ctr – control.
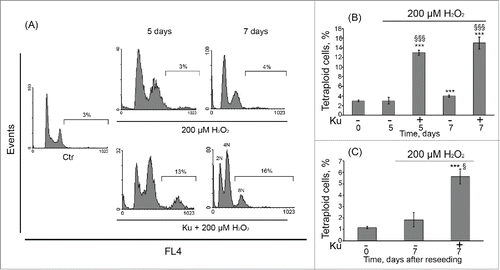
Tetraploidization of senescent hMESCs as a result of ATM kinase inhibition
Simultaneously with the accumulation of Ku-pretreated cells in G2/M phase, ATM kinase inhibition caused tetraploidization of H2O2-stimulated hMESCs, resulting in 8n DNA content. The tetraploid cell fraction gradually increased up to 16% within 7 d after the oxidative stress (). The FACS profile of these cells showed discrete peaks suggesting that the whole genome was duplicated. In order to elucidate the survival of the tetraploid cell fraction, Ku-treated cells were reseeded in 5 d after oxidative stress and additionally cultured for 7 d. As a result, we did not observe the disappearance of the tetraploid cell fraction in reseeded cells, suggesting their long-term viability (). Summarizing these findings, we can assume that ATM kinase inhibition allows H2O2-treated hMESCs to escape permanent cell cycle arrest due to loss of the functional ATM/p53/p21/Rb pathway however it causes the emergence of the tetraploid cells.
Figure 5. Tetraploidization of senescent hMESCs induced by Ku application. (A) hMESCs were left untreated (Ctr-control) or were treated either with H2O2 alone or in presence of the inhibitor and analyzed by FACS at indicated time points. The percentage of cells with DNA content > 4n is given. Representative FACS analyses are shown. (B) Quantification of tetraploidy induced by Ku. hMESCs were treated and analyzed as in (A). M ± SD, N = 3, ***p<0.005, versus control, §§§p<0.005, versus H2O2-treated cells, ns – not significant (Ctr – control). (C) Viability of the Ku-induced tetraploid cells. hMESCs were treated as in (A), then cells were reseeded in 5 d, additionally cultured for 7 d and analyzed by FACS. M ± SD, N = 3, ***p<0.005, versus control, §p<0.05, versus H2O2-treated cells. Ctr – control.
Ku blocks entry of H2O2-treated hMESCs into mitosis
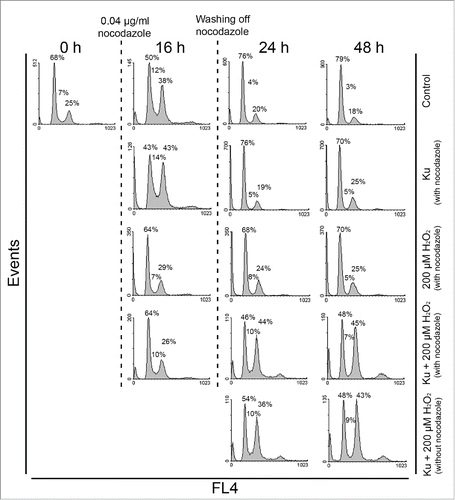
Having identified an increasing fraction of tetraploid cells in senescent hMESCs under Ku treatment, further we tried to enlighten the possible mechanism of tetraploidization. Theoretically, increased nuclear DNA amounts can result from endoreduplication cycles. Endoreduplication is a form of somatic polyploidy in which the chromosomes replicate time after time with no chromosome condensation and no mitosis. In favor of this assumption, we did not observe any metaphase cells bearing condensed chromosomes among the population of H2O2-treated hMESCs in presence of Ku using Giemsa banding (data not shown). The absence of mitotic cells allows us to suggest that they have become arrested in cell cycle prior to mitosis. To test this idea, we performed a set of experiments with the use of nocodazole (Noc). It is well known that Noc inhibits microtubule polymerization, thereby activating the spindle assembly checkpoint and preventing the cell exit from mitosis. The cell treatment with Noc for 16 h at a low concentration (0.04 μg/ml) resulted in accumulation of almost 40% of control (untreated) cells in G2/M phase without evident apoptosis. In 24 h after washing off Noc, more than 70% of control cells entered G0/G1 phase indicating the successful escape from Noc-induced G2/M block (). Of note, in response to Noc the cycle phase distribution of Ku-alone-treated cells and control cells were alike. In contrast to control hMESCs, H2O2-stimulated cells both in absence and presence of Ku were not able to accumulate in G2/M phase under Noc. Remarkably, (Ku+H2O2)-stimulated hMESCs accumulated in G2 phase of the cell cycle in 1 or 2 d after washing off Noc, and besides the phase distribution of these cells was similar to that without Noc treatment. The obtained data indicate the disturbance of G2-M transition, resulting in failure of Ku-treated senescent hMESCs to reach mitosis.
Figure 6. Cell cycle phase distribution of H2O2-treated hMESCs in presence of Ku was unaffected by Noc application. Control, Ku-, H2O2-, and (Ku+H2O2)-treated hMESCs were exposed to 0.04 μg/ml Noc for 16 h. After Noc treatment cells were either analyzed by FACS or washed and additionally cultured in the fresh medium with the following analysis in 24 or 48 h. To reveal the effect of Noc, (Ku+H2O2)-treated hMESCs without 16 h Noc application were analyzed at the same time points. The percentage of cells in G0/G1, S and G2/M is given. Representative FACS analyses are shown.
These observations were additionally verified by Western blot analysis with the use of specific antibody against phosphorylated histone H3 (Ser10). Due to the fact that site-specific phosphorylation of histone H3 at Ser10 tightly correlates with chromosome condensation and becomes maximal during metaphase of the mitosis, it is regarded as a reliable mitotic marker.Citation42 ATM inhibition completely abolished H3 prosphorylation in H2O2-treated hMESCs in comparison to control cells both in presence and absence of Noc (Fig. S1). Taking together, the results presented indicate that ATM down regulation, on the one hand, allows H2O2-treated hMESC to escape multiphase cell cycle arrest due to loss of the functional ATM/p53/p21/Rb pathway, and, on the other hand, it can induce bypass of mitosis and re-entry into S-phase, resulting in the emergence tetraploid cells.
Pifithrin-α triggers autophagy of H2O2-treated hMESCs
p53 was reported to be required to block entry of tetraploid cells into S phase and to suppress their proliferation.Citation43,44 So, one of the possible underlying causes of tetraploid cell emergence upon Ku treatment might be downregulation of p53 activity in senescent hMESCs. In order to check this suggestion, further we applied an inhibitor – Pifithrin-α (PFT) that specifically blocks transactivation of p53-responsive genes.Citation45 In the culture studies of p53 functions PFT is typically used at rather high concentration range from 30 up to 60 μM.Citation46,47 In our experimental conditions we decided to apply 50 μM PFT and showed that this concentration PFT had no effect on cell viability, decreased p21 protein levels and restored Rb phosphorylation in H2O2-treated hMESCs (). Unexpectedly, we did not observe any G2/M phase accumulation or tetraploidization in the population of (PFT+H2O2)-treated hMESCs () in contrast to (Ku+H2O2)-treated cells ().
Figure 7. (A) Application of 50 μM PFT had no effect on viability of H2O2-treated hMESCs. Cells were either pretreated or not with 50 μM PFT for 2 h, then subjected to 200 μM H2O2 for 1 h with following H2O2 replacement and cell cultivation under normal conditions. The percentage of viable cells was evaluated in 24 h after treatment by FACS analysis as described in “Materials and Methods” section. Results are shown as a percent of control. M ± SD, N = 3, *p<0.05, versus control. (B) Western blot analysis of p53 and Rb phospohorylation, as well as p21 protein expression. Representative results of the three experiments are shown in the figure. GAPDH was used as loading control. Ctr – control.
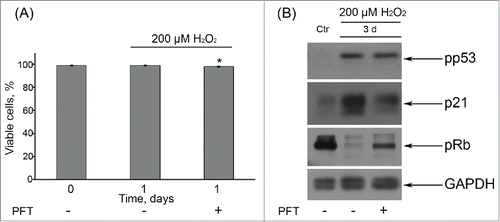
Figure 8. (A) p53 inhibition by PFT had no significant effect on the cell cycle phase distribution of H2O2-treated hMESCs. Flow cytometric analysis of cell cycle phase distribution: the percentage of cells in the G0/G1, S, and G2/M phases. M ± SD, N = 3, *p<0.05, ***p<0.005, versus control, <p<0.05, §§p<0.01, versus H2O2-treated cells. (Ctr – control). Representative FACS analyses are shown. (B) PFT-induced cell death of H2O2-treated hMESCs. Dead cells were determined at indicated time points after the treatment by FACS analysis as the percent of PI-positive cells. M ± SD, N = 3, *p<0.05, ***p<0.005, versus control, §§§p<0.005, versus H2O2-treated cells.
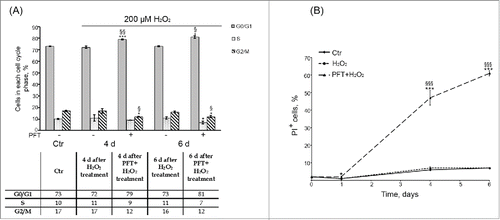
Moreover, PFT-induced inhibition of p53 transcriptional activity promoted a time-lag cell death of H2O2-treated cells, as at day 6 about 60% of (PFT+H2O2)-stimulated hMESCs were propidium iodide (PI)-positive (). We then performed a more specific kinetic analysis of H2O2-treated hMESCs death induced by PFT with the use of Annexin-V/PI (AnV/PI) assay. As shown in Figure S2, at day 3 the number of AnV positive cells (AnV+/PI- and AnV+/PI+) increased up to 13% in the population of (PFT+H2O2) cells. Notably, AnV is known to be an appropriate marker of both types of programmed cell death — apoptosis and autophagy — because cells undergoing either apoptosis or autophagy were shown to externalize phosphatidylserine residues, which normally are located on the internal surface of the plasma membrane.Citation48 At the same time we did not reveal activation of initiator caspases-8,-9 or effector caspase-3 (data not shown), that are typical of apoptosis.Citation49 This observation led us to investigate autophagy as a potential mechanism of PFT-induced cell death. In favor of this assumption with we detected approximately a 5-fold PFT-induced increase in side scatter (SS) at day 6 after the oxidative stress (). The SS elevation reflects an increase in internal granularity, corresponding to the increased autophagosome formation.Citation50
Figure 9. PFT promoted an internal granularity increase in H2O2-treated hMESCs. (A) Cells were treated as indicated in legend to Figure 8. Cell granularity was determined at indicated time points as the side scatter (SS) alterations. M ± SD, N = 3, *p<0.05, §§§ p<0.005, versus control, §§§p<0.005, versus H2O2-treated cells. Ctr – control. (B) Typical presentation of SS, reflecting the internal granularity of the cells. Data were obtained by light-scattering cytometry with using Win MDI program version 2.8.
These results were further expanded by the LC3-II levels monitoring at different time points after the oxidative stress. Being a pivotal component of the autophagic machinery LС3 is converted to lipidated LC3 (LC3-II) after autophagy induction. Immunoblotting of LC3 most commonly reveals 2 bands: LC3-I (18 kDa) and LC3-II (16 kDa). The LC3-II amount correlates well with the number of autophagosomes.Citation51 clearly shows that selective inhibition of p53 by PFT resulted in the appearance of a distinct LC3-II band at day 4 after H2O2 treatment that persisted up to day 7. These data support the hypothesis of autophagy initiation in H2O2-stimulated hMESCs upon PFT.
Figure 10. p53 down regulation by PFT caused the emergence of the typical autophagy markers in H2O2-treated hMESCs. (A) Cells were treated as indicated in the legend to Figure 7 and the relative abundance of LC3-I and LC3-II was determined by immunoblot with the use of specific antibodies. (B) PFT-dependent phosphorylation of Ulk1 at Ser555 in H2O2-treated cells was analyzed by Western blotting at the indicated time points. Representative results of the three experiments are shown in the figure. GAPDH was used as loading control. Ctr –control.
Another important protein involved in the initiation of autophagosome formation is Unc-51-like kinase 1 (Ulk1).Citation52 Previously it has been demonstrated that adenosine monophosphate-activated protein kinase (AMPK) activates autophagy by Ser555 phosphorylation of Ulk1Citation53, thus we analyzed the Ulk1 phosphorylation levels upon p53 inhibition in H2O2-treated hMESCs. Indeed, already in 2 d after the oxidative stress we revealed an enhanced phosphorylation of Ulk1 at Ser555 in PFT-treated cells compared to H2O2-stimulated cells (). Summarizing the obtained results, we conclude that the major cause of stressed cells’ death induced by p53 inhibition was not apoptosis, but autophagy.
Discussion
In the present study we investigated the effects of ATM as well as p53 down-regulation on the oxidative stress-induced hMESCs senescence development. Earlier we discovered that hMESCs treatment with sublethal H2O2 resulted in a rapid ATM activation that persisted for several days, suggesting its role in both initiation and stabilization of the premature senescence in hMESCs.Citation23 It is well documented that ATM is involved in various cellular stress responses, including cell cycle arrest, senescence and apoptosis.Citation24,25 Using a specific inhibitor of ATM activity – Ku, firstly we estimated ATM contribution to H2O2-induced senescence of hMESCs. ATM down regulation did not prevent the premature senescence of hMESCs, as Ku treatment had no effect on either proliferation restoration or the main H2O2-induced phenotypic changes, in particular increased cell size and enhanced SA-β-Gal staining. In contrast to our findings, recent reports claimed that Ku-induced ATM inhibition prevented the oxidative stress-induced senescence of biliary epithelial cells,Citation54 as well as H2O2-or high glucose-induced senescence of human umbilical vein endothelial cells.Citation14,55 However we cannot agree with the authors that a decrease in SA-β-Gal staining without any correlation to the cell proliferation recovery may serve as a convincing argument to postulate the complete senescence reversal. In case of hMESCs, we suppose that in the absence of functional ATM kinase another sensor kinase - ATR might be responsible for H2O2-induced senescence development. In favor of this assumption, it was shown that dual downregulation of both kinases by either CGK733 – a selective inhibitor of ATM/ATR, or siRNA-mediated knockdown of ATM/ATR promoted the proliferation and thus significantly increased lifespan of normal human cells.Citation27
Our previous data clearly demonstrated that hMESCs subjected to sublethal oxidative stress displayed the multiphase cell cycle arrest with preferable accumulation of cells in G1 phase.Citation41 As ATM kinase commonly via Chk2/p53/p21 pathway mediates G1/S cell cycle checkpoint, which prevents cells with damaged DNA from entering S-phase,Citation56 theoretically ATM down regulation should abolish this checkpoint. Indeed, after Ku treatment we revealed a prolonged accumulation of H2O2-stimulated hMESCs in G2/M phase of the cell cycle. Likewise, IR-treated normal human fibroblasts and etoposide-treated SW620 tumor cells also were arrested in G2 phase in response to ATM inhibition.Citation29 DNA damaged A-T cells lacking active ATM were also shown to be blocked at the G2 phase of cell cycle.Citation57 By contrast, Ku application led to the abolishment of G2 phase arrest in IR-exposed human embryonic stem cells.Citation58 Interestingly, dental pulp and periodontal ligament human mesenchymal stem cells subjected to IR were characterized by the ATM-independent G2 arrest.Citation34 We can assume that observed differences in the cell cycle phase distribution under ATM inhibition are associated with the cell specificity.
According to modern concepts, there are several consequences of a prolonged cell accumulation in G2 phase. One is a so-called checkpoint adaptation, meaning cycle re-entering following prolonged G2 accumulation in presence of irreparable DNA damage, thus transmitting damaged DNA to daughter cells.Citation59 Previously this phenomenon was described mainly in human cancer cells,Citation59,60 however further similar results were obtained in non-transformed human cells.Citation61,62,63 Another outcome of G2 cell accumulation suggests bypass of mitosis and re-entry into S phase resulting in tetraploid cells.Citation44 In favor of this statement, we observed the emergence of the tetraploid cell fraction in the population of Ku-treated hMESCs that increased in time and persisted even if cells were reseeded, suggesting their long-term viability. Based on the presented findings we conclude that ATM inhibition firstly allowed H2O2-treated hMESCs to escape permanent cell cycle arrest due to down regulation of the ATM/p53/p21/Rb pathway, and secondly prevented damaged cells from the mitosis entry and caused the emergence of the tetraploid cells.
Taking into account that suppression of the ATM activity did not lead to the desired elimination of the senescent cells from the hMESCs population, further we focused on the investigation of p53 inhibition impact. Earlier it was postulated that various tumor suppressor genes, including p53 actively repress tetraploidy because their removal can either stimulate the spontaneous tetraploidization of cells or facilitate the tetraploid cells’ survival.Citation44,64,65 Therefore, we hypothesized that p53 down regulation may underlie the observed tetraploidization of hMESCs. Contrary to our assumptions, p53 activity suppression caused a postponed and pronounced death of H2O2-treated hMESCs via autophagy. Autophagy is defined as an evolutionary conserved catabolic pathway by which cellular proteins and organelles are delivered to lysosomes for degradation and recycling.Citation66,67 Previous reports postulated a dual role of p53 in the autophagy regulation: it is capable both to enhance and to repress autophagy.Citation36,66 Our data confirm the results obtained on the cultures of various mammalian cells, indicating that p53 inactivation triggered autophagy.Citation36,37,38
In conclusion, hMESCs outline an easily available source for tissue regeneration due to their multilineage differentiation capacity, immunomodulatory properties and non-invasive isolation procedures.Citation39,40 Presently, there are the positive clinical results in hMESCs-based therapies of heart failure, Duchenne muscular dystrophy, type I diabetes, infertility.Citation68,69,70 Being transplanted hMESCs often get into the oxidative stress conditions which accompany the progression of many diseases what potentially may lead to loss of their regenerative potential and to the premature senescence. The findings presented in this study imply that continuously activated ATM is essential for genomic stability of the senescent hMESCs, as ATM down regulation causes the emergence of tetraploid cells. On the contrary, senescent hMESCs death occurred only under p53 suppression, therefore not ATM, but p53 down regulation could be considered as an effective tool for the elimination of the senescent cells from the population of H2O2-stimulated hMESCs.
Materials and methods
Cell culture
Human mesenchymal stem cells were isolated from desquamated endometrium in menstrual blood from healthy donors (hMESCs, line 2304) as described previously.Citation71 hMESCs have a positive expression of CD73, CD90, CD105, CD13, CD29, and CD44 markers and absence of expression of the hematopoietic cell surface antigens CD19, CD34, CD45, CD117, CD130, and HLA-DR (class II). Multipotency of isolated hMESCs is confirmed by their ability to differentiate into other mesodermal cell types, such as osteocytes and adipocytes. Besides, the isolated hMESCs partially (over 50%) express the pluripotencymarker SSEA-4 but do not express Oct-4. Immunofluorescent analysis of the derived cells revealed the expression of the neural precursor markers nestin and beta-III-tubulin. This suggests a neural predisposition of the established hMESCs. These cells are characterized by high rate of cell proliferation (doubling time 22–23 h) and high cloning efficiency (about 60%). hMESCs were cultured in complete medium DMEM/F12 (Gibco BRL, USA) supplemented with 10% FBS (HyClone, USA), 1% penicillin-streptomycin (Gibco BRL, Gaithersburg, MD, USA)and 1% glutamax (Gibco BRL, USA) at 37˚C in humidified incubator, containing 5% CO2. Cells were harvested by trypsinization and plated at a density of 15*10Citation3 cells per cm2. To avoid complications of replicative senescence, cells at early passages (between 5 and 9 passages) were used in all experiments.
Cell treatments
H2O2 treatment was performed on the subconfluent cells to avoid variability of H2O2 toxicity. H2O2 stock solution in serum-free medium was prepared from 30% H2O2 (Sigma, USA) just before adding. Cells were treated with 200 μM H2O2 for 1 h, then washed twice with serum-free medium to remove H2O2, and re-cultured in fresh complete medium for various durations as specified in individual experiments. To inhibit ATM kinase activity, 10 μM Ku55933 dissolved in DMSO (Merk, Germany) was added in the culture medium 40 min before H2O2-treatment. To suppress p53 activity, cells were treated with 50 μM Pifithrin-α (Merk, Germany) during 2 h before H2O2 treatment. Both inhibitors were presented in the culture medium during oxidative stress and then have been added daily to avoid their degradation. In the set of experiments with nocodazole, cells were either pretreated or not with Ku for 40 min, followed by H2O2 stimulation for 1 h, and then treated with 0.04 μg/ml nocodazole for 16 h.
Assessment of the cell viability
The cell viability after incubation with the inhibitor was evaluated by the enzymatic conversion of MTT (AppliChem, Germany) to formazan in live cells. The culture medium from the cells grown in plates was removed, and 3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT; 0,715mg/mL) in serum-containing growth medium was added to each well. In 2 h the solution was changed to DMSO to solve formazan produced. The plates were shaken for 15 min at room temperature; thereafter the absorbance was measured at 570 nm using microplate reader (Fluorofot “Charity,”Russia). All points were read as parallels of 8 similar samples. The average absorbance at a given time point was normalized to the start time point.
FACS analysis
FACS analysis for DNA content was performed using standard procedures and PI DNA staining as described previously.Citation23 Flow cytometry was performed using the Coulter EPICS XL Flow Cytometer (Backman Coulter, USA) and the obtained data were analyzed using Win MDI software version 2.8. The cell size was evaluated by cytometric light scattering of PI-stained cells. To discriminate the live and dead cells, 2-parameter histogram was used (FL4LOG vs. FSLOG). Analysis of each sample (at least 10,000 cells) was performed for 100 sec with high sample delivery. Analysis of apoptotic cells was performed using a FITC-annexin V staining according to standard manufacture protocols (BD Pharmingen, USA).
Western blotting
Western blot analysis was performed as described previously.Citation72 SDS-PAGE electrophoresis, transfer to nitrocellulose membrane and immunoblotting with ECL (Thermo Scientific, USA) detection were performed according to standard manufacturer's protocols (Bio-Rad Laboratories, USA). Antibodies against the following proteins were used: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (clone 14C10), (1:1000, #2118, Cell Signaling, USA), phospho-ATM (Ser1981) (clone D6H9) (1:1000, #5883, Cell Signaling, USA), phospho-Chk2 (Thr68) (1:1000, #2661, Cell Signaling, USA), phospho-p53 (Ser15) (clone 16G8) (1:1000, #9286, Cell Signaling, USA), p21Waf1/Cip1 (clone 12D1) (1:1000, #2947, Cell Signaling, USA), phospho-Rb (1:1000, Ser807/811) (#9308, Cell Signaling, USA), phospho-Histone H3 (Ser10) (1:1000, #9706, Cell Signaling, USA), LC3 (1:500, #ABC232, Merk KGaA, Germany), phospho-Ulk1 (Ser555) (1:5000, #ABC124, Merk KGaA, Germany), as well as horseradish peroxidase-conjugated goat anti-rabbit IG (1:10000, #7074, Cell Signaling, USA) or horseradish peroxidase-conjugated goat anti-mouse IG (1:10000, #7076, Cell Signaling, USA). Hyperfilm (CEA) was from Amersham (Sweden). Equal protein loading was confirmed by Ponceau S (Sigma, USA) staining.
Statistics
All data are presented as the mean and standard deviation of the mean from at least 3 separate experiments performed. Statistical differences were calculated using the Student's t-test and considered significant at *,§ p < 0.05; **,§§ p < 0.01; ***, §§§ p < 0.005.
Abbreviations
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| SA-β-Gal | = | senescence-associated β-galactosidase |
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| FACS | = | flow-activated cell sorting |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
Abbreviations
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| SA-β-Gal | = | senescence-associated β-galactosidase |
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| FACS | = | flow-activated cell sorting |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
Abbreviations
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| SA-β-Gal | = | senescence-associated β-galactosidase |
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| FACS | = | flow-activated cell sorting |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
Abbreviations
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| SA-β-Gal | = | senescence-associated β-galactosidase |
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| FACS | = | flow-activated cell sorting |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
Abbreviations
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| SA-β-Gal | = | senescence-associated β-galactosidase |
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| FACS | = | flow-activated cell sorting |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
Abbreviations
| ATM | = | ataxia-telangiectasia mutated |
| DDR | = | DNA damage response |
| FACS | = | flow-activated cell sorting |
| hMESCs | = | human endometrium-derived mesenchymal stem cells |
| Noc | = | nocodazole |
| PFT | = | pifithrin-α |
| SA-β-Gal | = | senescence-associated β-galactosidase |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
New_Suppl_Files.zip
Download Zip (459.1 KB)KCCY_S_1121326.zip
Download Zip (449.8 KB)Funding
A. Borodkina, E. Burova, N. Nikolsky, A. Shatrova and N. Grukova acknowledge the support from the Russian Science Foundation (project №14-50-00068) with the financial support from the Federal Agency of Scientific Organizations (Russia). E. Burova and P. Deryabin acknowledge the support from the Russian Foundation of Basic Research (project №14-04-01720).
Related Research Data
References
- Wang D, Jang DJ. Protein kinase CK2 regulates cytoskeletal reorganization during ionizing radiation-induced senescence of human mesenchymal stem cells. Cancer Res 2009; 69:8200-7; PMID:19826041; http://dx.doi.org/10.1158/0008-5472.CAN-09-1976
- Brandl A, Meyer M, Bechmann V, Nerlich M, Angele P. Oxidative stress induces senescence in human mesenchymal stem cells. Exp Cell Res 2011; 317:1541-7; PMID:21376036; http://dx.doi.org/10.1016/j.yexcr.2011.02.015
- Kim JS, Kim EJ, Kim HJ, Yang JY, Hwang GS, Kim CW. Proteomic and metabolomic analysis of H2O2-induced premature senescent human mesenchymal stem cells. Exp Gerontol 2011; 46:500-10; PMID:21382465; http://dx.doi.org/10.1016/j.exger.2011.02.012
- Cmielova J, Havelek R, Soukup T, Jiroutova A, Visek B, Suchanek J, Vavrova J, Mokry J, Muthna D, Bruckova L, et al. Gamma radiation induces senescence in human adult mesenchymal stem cells from bone marrow and periodontal ligaments. Int J Radiat Biol 2012; 88:393-404; PMID:22348537; http://dx.doi.org/10.3109/09553002.2012.666001
- Larsen SA, Kassem M, Rattan SI. Glucose metabolite glyoxal induces senescence in telomerase-immortalized human mesenchymal stem cells. Chem Cent J 2012; 6:18; PMID:22424056; http://dx.doi.org/10.1186/1752-153X-6-18
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/10.1038/nrm2233
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging 2012; 4:159-65; PMID:22394614
- Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G, Galderisi U, Chambery A. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis 2013; 4:e911; PMID:24201810; http://dx.doi.org/10.1038/cddis.2013.445
- Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013; 12:489-98; PMID:23521863; http://dx.doi.org/10.1111/acel.12075
- Harley CB, Vaziri H, Counter CM, Allsopp RC. The telomere hypothesis of cellular aging. Exp Gerontol 1992; 27:375-82; PMID:1459213; http://dx.doi.org/10.1016/0531-5565(92)90068-B
- Toussaint O, Remacle J, Dierick JF, Pascal T, Frippiat C, Zdanov S, Magalhaes JP, Royer V, Chainiaux F. From the Hayflick mosaic to the mosaics of ageing. Role of stress-induced premature senescence in human ageing. Int J Biochem Cell Bio 2002; 34:1415-29; PMID:12200036; http://dx.doi.org/10.1016/S1357-2725(02)00034-1
- Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest 2004; 113:8-13; PMID:14702100; http://dx.doi.org/10.1172/JCI200420663
- Ksiazek K, Passos JF, Olijslagers S, Saretzki G, Martin-Ruiz C, von Zglinicki T. Premature senescence of mesothelial cells is associated with non-telomeric DNA damage. Biochem Biophys Res Commun 2007; 362:707-11; PMID:17720141; http://dx.doi.org/10.1016/j.bbrc.2007.08.047
- Zhan H, Suzuki T, Aizawa K, Miyagawa K, Nagai R. Ataxia Telangiectasia Mutated–mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem 2010; 285:29662-70; PMID:20639198; http://dx.doi.org/10.1074/jbc.M110.125138
- Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 2012; 37:15-22; PMID:22079189; http://dx.doi.org/10.1016/j.tibs.2011.10.002
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorilation and dimer dissociation. Nature 2003; 421:499-506; PMID:12556884; http://dx.doi.org/10.1038/nature01368
- So S, Davis AJ, Chen DJ. Autophosphorilation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol 2009; 187:977-990; PMID:20026654; http://dx.doi.org/10.1083/jcb.200906064
- Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene 2003; 22:9030-40; PMID:14663481; http://dx.doi.org/10.1038/sj.onc.1207116
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997; 277:831-4; PMID:9242615; http://dx.doi.org/10.1126/science.277.5327.831
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/10.1101/gad.13.12.1501
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008; 8:512-22; PMID:18574463; http://dx.doi.org/10.1038/nrc2440
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence Mol Syst Biol 2010; 6:347; PMID:20160708; http://dx.doi.org/10.1038/msb.2010.5
- Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 2014; 6:481-95; PMID:24934860
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9:759-69; PMID:18813293; http://dx.doi.org/10.1038/nrm2514
- Sullivan KD, Gallant-Behm CL, Henry RE, Fraikin JL, Espinosa JM. The p53 circuit board. Biochim Biophys Acta 2012; 1825:229-44; PMID:22333261
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Stephen P. Jackson SP. A DNA damage checkpoint response in telomere- initiated senescence. Nature 2003; 426:194-8; PMID:14608368; http://dx.doi.org/10.1038/nature02118
- Won J, Kim M, Kim N, Ahn JH, Lee WG, Kim SS, Chang KY, Yi YW, Kim TK. Small molecule–based reversible reprogramming of cellular lifespan. Nat Chem Biol 2006; 2:369-74; PMID:16767085; http://dx.doi.org/10.1038/nchembio800
- Cao L, Kawai H, Sasatani M, Iizuka D, Masuda Y, Inaba T, Suzuki K, Ootsuyama A, Umata T, Kamiya K1, Suzuki F. A novel ATM/TP53/p21-mediated checkpoint only activated by chronic γ-irradiation. PLoS One 2014; 9:e104279; PMID:25093836; http://dx.doi.org/10.1371/journal.pone.0104279
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP,Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152-9; PMID:15604286; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727
- Crescenzi E, Palumbo G, de Boer J, Brady HJM. Ataxia Telangiectasia Mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: implications for chemotherapy. Clin Cancer Res 2008; 14:1877-87; PMID:18347191; http://dx.doi.org/10.1158/1078-0432.CCR-07-4298
- Alao JP, Sunnerhagen P. The ATM and ATR inhibitors CGK733 and caffeine suppress cyclin D1 levels and inhibit cell proliferation. Radiat Oncol 2009; 4:51; PMID:19903334; http://dx.doi.org/10.1186/1748-717X-4-51
- Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, Eckhardt SG, Tan AC, DeGregori J, Espinosa JM. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol 2012; 8:646-54; PMID:22660439; http://dx.doi.org/10.1038/nchembio.965
- Zhang X, Li J, Sejas DP, Pang Q. The ATM/p53/p21 pathway influences cell fate decision between apoptosis and senescence in reoxygenated hematopoietic progenitor cells. J Biol Chem 2005; 280:19635-40; PMID:15753076; http://dx.doi.org/10.1074/jbc.M502262200
- Cmielova J, Havelek R, Kohlerova R, Soukup T, Bruckova L, Suchanek J, Vavrova J, Mokry J, Rezacova M. The effect of ATM kinase inhibition on the initial response of human dental pulp and periodontal ligament mesenchymal stem cells to ionizing radiation. Int J Radiat Biol 2013; 89:501-11; PMID:23425510; http://dx.doi.org/10.3109/09553002.2013.775530
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 path ways. EMBO J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/10.1093/emboj/cdg417
- Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. Adualroleofp53in thecontrolofautophagy. Autophagy 2008; 4:810-4; PMID:18604159; http://dx.doi.org/10.4161/auto.6486
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Bio 2010; 22:181-5; PMID:20044243; http://dx.doi.org/10.1016/j.ceb.2009.12.001
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nature Cell Biol 2008; 10:676-87; PMID:18454141; http://dx.doi.org/10.1038/ncb1730
- Meng X, Ichim TE, Zhong J, Rogers A, Yin Z, Jackson J, Wang H, Ge W, Bogin V, Chan KW, et al. Endometrial regenerative cells: A novel stem cell population. J Transl Med 2007; 5:57; PMID:18005405; http://dx.doi.org/10.1186/1479-5876-5-57
- Patel AN, Park E, Kuzman M, Benetti F, Silva FJ, Allickson JG. Multipotent menstrual blood stromal stem cells: isolation, characterization, and differentiation. Cell Transpl 2008; 17:303-11; PMID:18522233; http://dx.doi.org/10.3727/096368908784153922
- Burova E, Borodkina A, Shatrova A, Nikolsky N. Sublethal oxidative stress induces the premature senescence of human mesenchymal stem cells derived from endometrium. Oxid Med Cell Longev 2013; 474931; PMID:24062878
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997; 106:348-60; PMID:9362543; http://dx.doi.org/10.1007/s004120050256
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12:1315-28; PMID:11359924; http://dx.doi.org/10.1091/mbc.12.5.1315
- Davoli T, Denchi EL, Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141:81-93; PMID:20371347
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285:1733-7; PMID:10481009; http://dx.doi.org/10.1126/science.285.5434.1733
- Wan C, Ma X, Shi S, Zhao J, Nie X, Han J, Xiao J, Wang X, Jiang S, Jiang J. Pivotalrolesofp53transcription-dependentand -independentpathwaysinmanganese inducedmitochondrial dysfunctionandneuronalapoptosis. Toxicol Appl Pharmacol 2014; 281:294-302; PMID:25448048; http://dx.doi.org/10.1016/j.taap.2014.10.013
- Yeh YC, Liu TJ, Lai HC. Shikonin induces apoptosis, necrosis, and premature senescence of human A549 lung cancer cells through upregulation of p53 expression. Evid Based Complement Alternat Med 2015; 2015:620383; PMID:25737737
- Gosselin K, Deruy E, Martien S, Vercamer C, Bouali F, Dujardin T, Slomianny C, Houel-Renault L, Chelli F, De Launoit Y et al. Senescent keratinocytes die by autophagic programmed cell death. Am J Pathol 2009; 174:423-35; PMID:19147823; http://dx.doi.org/10.2353/ajpath.2009.080332
- Kitazumi I, Tsukahara M. Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J 2011; 278:427-41; PMID:21182594; http://dx.doi.org/10.1111/j.1742-4658.2010.07975.x
- Suárez AL, Kong R, George T, He L, Yue Z, van Dyk LF. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J Virol 2011; 85:6293-308; PMID:21490089; http://dx.doi.org/10.1128/JVI.00001-11
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Chen Y, Klionsky DJ. The regulation of autophagy – unanswered questions. J Cell Sci 2011; 124:161-70; PMID:21187343; http://dx.doi.org/10.1242/jcs.064576
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Sasaki M, Ikeda H, Sato Y, Nakanuma Y. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of ATM pathway: a culture study. Free Radic Res 2008; 42:625-32; PMID:18608517; http://dx.doi.org/10.1080/10715760802244768
- Xu TY, Ding QF, Wan ZH, Zeng L, Chang BB, Zhang XJ. The role of DNA double strains damage repairing mechanisms in high glucose-induced endothelial senescence. Sichuan Da Xue Xue Bao Yi Xue Ban 2011; 42:190-3; PMID:21500551
- Weber AM, Ryan AJ. ATMandATRastherapeutictargetsincancer. Pharmacol Ther 2015; 149:124-38; PMID:25512053; http://dx.doi.org/10.1016/j.pharmthera.2014.12.001
- Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G2-M checkpoints are induced by ionizing irradiation. Mol Cell Biol 2002; 22:1049-59; PMID:11809797; http://dx.doi.org/10.1128/MCB.22.4.1049-1059.2002
- Momcilovic O , Choi S , Varum S , Bakkenist C , Schatten G , Navara C. Ionizing radiation induces ataxia telangiectasia mutated dependent checkpoint signaling and G(2) but not G(1) cell cycle arrest in pluripotent human embryonic stem cells. Stem Cells 2009; 27:1822-35; PMID:19544417; http://dx.doi.org/10.1002/stem.123
- Syljuasen RG. Checkpoint adaptation in human cells. Oncogene 2007; 26:5833-39; PMID:17384683; http://dx.doi.org/10.1038/sj.onc.1210402
- Kubara PM, Kernéis-Golsteyn S, Studény A, Lanser BB, Meijer L, Golsteyn RM. Human cells enter mitosis with damaged DNA after treatment with pharmacological concentrations of genotoxic agents. Biochem J.2012; 446:373-81; PMID:22686412; http://dx.doi.org/10.1042/BJ20120385
- Deckbar D, Birraux J, Krempler A, Tchouandong L, Beucher A, Walker S, Stiff T, Jeggo P, Löbrich M. ChromosomebreakageafterG2checkpointrelease. J Cell Biol 2007; 176:749-55; PMID:17353355; http://dx.doi.org/10.1083/jcb.200612047
- Lobrich M, Jeggo PA. The impact of a negligent G2 /M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 2007; 7:861-9; PMID:17943134; http://dx.doi.org/10.1038/nrc2248
- van Vugt MA, Yaffe MB. Cell cycle re-entry mechanisms after DNA damage checkpoints: giving it some gas to shut off the breaks! Cell Cycle 2010; 9:2097-101; PMID:20505366; http://dx.doi.org/10.4161/cc.9.11.11840
- Margolis RL. Tetraploidy and tumor development. Cancer Cell 2005; 8:353-4; PMID:16286243; http://dx.doi.org/10.1016/j.ccr.2005.10.017
- Vitale I, Senovilla L, Jemaà M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 2010; 29:1272-84; PMID:20186124; http://dx.doi.org/10.1038/emboj.2010.11
- Rufini A, Tucci P, Celardo I, Melino G. Senescenceandaging: thecriticalroles ofp53. Oncogene 2013; 32:5129-43; PMID:23416979; http://dx.doi.org/10.1038/onc.2012.640
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA 2010; 107:18511-6; PMID:20937856; http://dx.doi.org/10.1073/pnas.1006124107
- Ichim TE, Alexandrescu DT, Solano F, Lara F, Campion Rde N, Paris E, Woods EJ, Murphy MP, Dasanu CA, Patel AN, et al. Mesenchymal stem cells as anti-inflammatories: implications for treatment of Duchenne muscular dystrophy. Cell Immunol 2010; 260:75-82; PMID:19917503; http://dx.doi.org/10.1016/j.cellimm.2009.10.006
- Bockeria L, Bogin V, Bockeria O, Le T, Alekyan B, Woods EJ, Brown AA, Ichim TE, Patel AN. Endometrial regenerative cells for treatment of heart failure: a new stem cells in clinic. J Transl Med 2013; 11:56; PMID:23510656; http://dx.doi.org/10.1186/1479-5876-11-56
- Ulrich D, Muralitharan R, Gargett CE. Toward the use of endometrial and menstrual blood stem cells for cell-based therapies. Expert Opin Biol Ther 2013; 13:1387-400; PMID:23930703; http://dx.doi.org/10.1517/14712598.2013.826187
- Zemelko VI, Grinchuk TM, Domnina AP, Artzibasheva IV, Zenin VV, Kirsanov AA, Bichevaia NK, Korsak VS, Nikolsky NN. Multipotent mesenchymal stem cells of desquamated endometrium: Isolation, characterization, and application as a feeder layer for maintenance of human embryonic stem cells. Cell and Tissue Biol 2012; 6:1-11; PMID:22359950; http://dx.doi.org/10.1134/S1990519X12010129
- Borodkina AV, Shatrova AN, Pugovkina NA, Zemelko VI, Nikolsky NN, Burova EB. Different protective mechanisms of human embryonic and endometrium-derived mesenchymal stem cells under oxidative stress. Cell and Tissue Biol 2014; 8:11-21; PMID:25486783; http://dx.doi.org/10.1134/S1990519X14010040