
KEYWORDS:
Crosstalk between the epithelial and stromal compartment is essential to maintain tissue homeostasis. Normal tissue environment can suppress the ability of incipient neoplastic cells to survive and proliferate; as a consequence, tumor cells often acquire the ability to create a permissive microenvironment. So far, genetic and epigenetic changes of the epithelium have been implicated as likely primary determinants of carcinogenesis, while alterations of the underlying mesenchyme have been assumed to be only a reactive consequence of a stromal-epithelial co-evolution process. Increasing amount of evidence points to the ability of the microenvironment to acquire a pro-carcinogenic state independently of the presence of tumor cells. Alteration of the stromal microenvironment, such as the appearance of cancer-associated fibroblasts (CAFs), is emerging as an important event in tumor initiation as well as progression.Citation1
We previously showed that mesenchymal deletion of CSL, the key effector of “canonical” Notch signaling, endowed with an intrinsic repressive function, results in spontaneous multifocal keratinocyte tumor formation, preceded by CAF phenotype acquisition in both mouse and human stromal fibroblast.Citation2 Recently we discovered, in the same mouse and human cell system, that CAF activation is accompanied by stromal cell senescence and that the 2 events are genetically linked.Citation3
More specifically, deletion or silencing of the CSL gene in dermal fibroblasts induces cellular senescence and increased expression of senescence determinants, such as p15, p16, p21 and miR-34a. In parallel, CSL silencing also results in induction of the senescence messaging secretome (SMS) constituents, such as IL6, IL8, Pai-1 and MMPs. We also identified key senescence-effector genes, specifically p16, p21, miR-34a and IL6, together with a broad spectrum of CAF effector genes as direct targets of CSL transcriptional repression.
Two CSL binding sites on p21 promoter have adjacent p53 recognition sequences, raising the possibility of direct interaction between the 2 proteins. Indeed, we subsequently uncovered that CSL and p53 can physically interact and that CSL suppresses p53 activity, blocking its association to p300, a critical chromatin modifier and transcriptional activator.
Previous work has shown that stromal cell senescence can be associated with induction of SMS and CAF effector genes, yet whether or not the 2 events can be genetically dissociated remained unclear.Citation4 We therefore set out to test the impact of p53, a well-known determinant of cellular senescence, on CSL dependent senescence and CAF activation. We found that loss of p53 overcame the senescence driven by CSL knockdown. Surprisingly, concomitant CSL and p53 silencing also enhanced the expression of CAF effector genes, like Cox-2 and periostin, known to be involved in inflammation and macrophage recruitment. To assess the in vivo significance of these findings, we developed a mouse ear intradermal injection assay for cancer and stromal cell expansion by in vivo imaging of fluorescently labeled cells. Skin or head and neck squamous cell carcinoma (SCC) cells admixed with fibroblasts deficient for both CSL and p53 showed enhanced outgrowth, leukocyte/macrophage infiltration and angiogenesis in comparison with CSL alone.
In most cases, in situ epithelial lesions do not progress into an invasive cancer and additional genetic changes are needed for progression into a malignancy. The events governing this process are not fully understood, but most likely a tumor-permissive microenvironment is required.Citation5 Stromal changes associated with cell senescence are thought to play an important role in the increased incidence of age-related cancers.Citation6 However, this process may be more relevant in the initial phase of cancer development rather than at an advanced stage, the latter being characterized by increased fibroblast density and proliferation.Citation7 Concurring with this possibility, we found senescent stromal cells in human premalignant skin lesions, actinic keratosis (AK) and in situ SCC, while little or none could be detected in invasive SCCs. Stromal dermal fibroblasts underlying AK, in situ and invasive SCC showed CSL down-modulation, while p53 gene expression was down-modulated only in SCCs. Furthermore, relative to normal HDFs, SCC-derived CAFs showed lower levels of CSL. Importantly, CSL over-expression in these cells resulted in suppression of CAF markers and their ability to promote SCC cell proliferation in vivo. The SCC-derived CAFs were also found to have lower p53 levels compared to normal HDFs, with FGF-2 paracrine signaling as the probable culprit of p53 down-modulation.
Thus far 2 apparently conflicting cellular events were linked to CAF determination. On one hand, fibroblast senescence was implicated as an important factor in CAF phenotype acquisition, with the SMS secretome leading to inflammation and tumorigenesis. On the other hand, hyperproliferating CAFs causing tumor-associated fibrosis were observed surrounding malignant epithelial tumors.
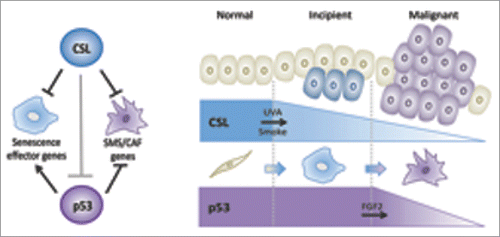
Our findings reconcile the 2 biological events and point to a multistep process of CAF activation under convergent CSL and p53 control. Activation of p53 provides a failsafe mechanism against consequences of compromised CSL activity in stromal cells. Concomitant p53 and CSL down-regulation are involved in subsequent stromal and cancer cell expansion ().
Figure 1. Combined CSL-p53 control of multistep CAF determination regulates tumor evolution. Left: CSL acts as a constitutive direct repressor of multiple senescence- and CAF-effector genes; it also physically interacts with p53 and represses its activity. Concomitant p53 and CSL loss in HDFs blocks senescence and enhances CAF effector gene expression. Right: Firstly, environmental insults like UVA irradiation and smoke can cause CSL down-modulation and stromal cell senescence. Secondly, paracrine influence of incipient cancer cells, like FGF-2 secretion, is involved in p53 suppression leading to stromal and tumor cell expansion.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Goruppi S, et al. Trends Cell Biol 2013; 23:593-602; PMID:24074947; http://dx.doi.org/10.1016/j.tcb.2013.08.006
- Hu B, et al. Cell 2012; 149:1207-20; PMID:22682244; http://dx.doi.org/10.1016/j.cell.2012.03.048
- Procopio MG, et al. Nat Cell Biol 2015; 17:1193-204; PMID:26302407; http://dx.doi.org/10.1038/ncb3228
- Alspach E, et al. Crit Rev Oncog 2013; 18:549-58; PMID:24579734; http://dx.doi.org/10.1615/CritRevOncog.2014010630
- Dotto GP. J Clin Invest 2014; 124:1446-53; PMID:24691479; http://dx.doi.org/10.1172/JCI72589
- Campisi J. Curr Opin Genet Dev 2011; 21:107-12; PMID:21093253; http://dx.doi.org/10.1016/j.gde.2010.10.005
- Paulsson J, et al. Semin Cancer Biol 2014; 25:61-8; PMID:24560651; http://dx.doi.org/10.1016/j.semcancer.2014.02.006