
ABSTRACT
We previously reported that aberrant TGF-β/Smad2/3 signaling in endometrial cancer (ECA) leads to continuous ubiquitylation of p27kip1(p27) by the E3 ligase SCF-Skp2/Cks1 causing its degradation, as a putative mechanism involved in the pathogenesis of this cancer. In contrast, normal intact TGF-β signaling prevents degradation of nuclear p27 by SCF-Skp2/Cks1 thereby accumulating p27 to block Cdk2 for growth arrest. Here we show that in ECA cell lines and normal primary endometrial epithelial cells, TGF-β increases Cdh1 and its binding to APC/C to form the E3 ligase complex that ubiquitylates Cks1 and Skp2 prompting their proteasomal degradation and thus, leaving p27 intact. Knocking-down Cdh1 in ECA cell lines increased Skp2/Cks1 E3 ligase activity, completely diminished nuclear and cytoplasmic p27, and obviated TGF-β-mediated inhibition of proliferation. Protein synthesis was not required for TGF-β-induced increase in nuclear p27 and decrease in Cks1 and Skp2. Moreover, half-lives of Cks1 and Skp2 were extended in the Cdh1-depleted cells. These results suggest that the levels of p27, Skp2 and Cks1 are strongly or solely regulated by proteasomal degradation. Finally, an inverse relationship of low p27 and high Cks1 in the nucleus was shown in patients in normal proliferative endometrium and grade I-III ECAs whereas differentiated secretory endometrium showed the reverse. These studies implicate Cdh1 as the master regulator of TGF-β-induced preservation of p27 tumor suppressor activity. Thus, Cdh1 is a potential therapeutic target for ECA and other human cancers showing an inverse relationship between Cks1/Skp2 and p27 and/or dysregulated TGF-β signaling.
Introduction
Aberrant signaling of the potent endogenous inhibitor of proliferation, transforming growth factor-β (TGF-β), occurs early in the oncogenesis of many human cancers including type I endometrial cancer (ECA).Citation1-5 ECA is the most common gynecological cancer occurring at a rate of approximately 54,870 in 2013 in the United States.Citation6 Two transmembrane receptors, TβRI and TβRII form a tetrameric complex with activated mature TGF-β ligand to transduce signaling by directly phosphorylating downstream transcription factors, Smad2 and Smad3, via TβRI serine/threonine kinase activity.Citation7 Activated Smad2 or Smad3 bind Smad4, translocate to the nucleus, and combine with DNA binding protein complexes on different promoter response elements for a plethora of activator or repressor activities.Citation8,9 TGF-β mediates cell cycle arrest in late G1 phase of the cell cycle through cell-type and context-dependent pathways and mechanisms.Citation10,11 Cyclins bind to specific Cdks that phosphorylate pRB for progression to S phase; this activity is blocked by cyclin-dependent kinase inhibitors (CDKIs).Citation12 Inhibition of proliferation can be mediated by the upregulation of 2 families of mammalian CDKIs, ink4 proteins, p15, p16, p18, p19 and kip/cip proteins, p21cip1, p27kip1, and p57, which act by blocking Cdk2/4/6 kinase activity. Importantly, TGF-β activates transcription of p15 and p21 which bind Cyclin D/Cdk4/6 promoting the binding of p27 from Cyclin D/Cdk4/6 to CyclinE/Cdk2 to block Cdk2 activity.Citation13 TGF-β also promotes the binding of p27 to CyclinE/Cdk2 to block pRb phosphorylation.Citation14 Another significant means for TGF-β to achieve growth inhibition is by downregulation of Myc transcription by the binding of Smad3/4, E2F4 and p107 to a TGF-β inhibitory element in the Myc promoter thereby reducing the expression of Myc targeted growth promoting genes.Citation15 Interestingly, whereas Smad7 is inhibitory by blocking Smad2/3-induced functions, TGF-β signaling can induce its cytostatic effect through ubiquitin-mediated degradation of Myc by Smad7 via the recruitment of the E3 ligase Skp2.Citation16
In addition to being under transcriptional and translational control, the levels of cell cycle proteins are precisely regulated by waves of ubiquitin-mediated degradation that oscillate with peaks in the levels of ubiquitin E3 ligases of the ubiquitin-proteasome system (UPS).Citation17,18 Two major multi-subunit E3 ligases that regulate cell cycle traverse are the Anaphase Promoting Complex/Cyclosome (APC/C) and the SCF-Skp2/Cks1 complex.Citation19 These E3 ligases cause degradation of cyclin/Cdks and their CDKIs in perfect synchrony to regulate cell cycle progression and arrest. Three enzymes (E1, E2, E3) collaborate to ultimately transfer/activate (E1), conjugate (E2) and ligate (E3) chains of ubiquitin to the target protein.Citation17 The E3 ligases provide substrate recognition and ubiquitylate their substrates for degradation by proteasomes. The level of the SCF-Skp2/Cks1 is high in G1/S causing the degradation of p27 to enable cell cycle progression.Citation20 APC specific E3 ligase activity is dependent on its binding to either Cdh1 or Cdc20, as catalytic co-activators of the APC/C.Citation21-23 APC binding to Cdc20 in late G2/early mitosis has E3 ligase specificity for securins and cyclins A and B and other cell cycle proteins involved in cell cycle progression whereas in late mitosis/early G1, Cdh1 displaces Cdc20 from the APC. APC/CCdh1 has substrate ubiquitylating specificity for Skp2 and Cks1 and other cell cycle proteins including Cdc20, causing their degradation in G0/G1 leaving p27 intact to effectuate G1 arrest.Citation24-27 The APC/CCdh1 complex, composed of 13 different subunit proteins termed Apc1-13,Citation28 is involved in controlling differentiation, genomic stability, and tumor suppression.Citation19,29-31 Inhibitors of the APC/C include Emi1/2, Bub3, and the mitotic checkpoint complex (MCC).Citation19
Whereas SCF-Skp2 complexed with different binding partners has substrate specificity for both tumor suppressors and oncogenes, uniquely, a pocket is formed by the binding of Cks1 (9.8 kDa) at the C-terminus of Skp2 (45 kDa) allowing substrate specificity for the CDKIs (tumor suppressors), p27 and p21.Citation32,33 Specific amino acid residues in Cks1 interact with p27 phosphorylated on T187 and the ubiquitylation of p27 by Skp2 ensues.Citation34-36 The presence of Cks1 in the SCF complex is rate limiting for p27 degradation.Citation37 Notably, apart from its adaptor role with the SCF-Skp2 complex, Cks1 has other important cellular functions that have been associated with increased proliferation and cancer including a plethora of intricate and complex cell cycle regulatory activities, one, being the regulation of APC/C and spindle assembly checkpoint for mitotic timing.Citation29,38-42 In addition, Cks1 has been shown to be involved in dephosphorylating Cdk1,Citation43 the recruitment of CyclinA/Cdc20 to phosphorylated APC/C for its ubiquitylation and degradation,Citation44,45 and in chromatin remodeling on the Cdc20 promoter.Citation46 Cks1 has several sites for physical protein-protein interaction including: the C-terminus of Cdk2, the C-terminal tail of Skp2, and hypothetically, p27-T187 within the concave groove formed by Skp2 interacting with Cks1, and the Cdc20 promoter;Citation33-35,41,47,48 all playing a role in the outcome of p27 functionality in cell cycle arrest.
We reported that perpetual ubiquitin mediated degradation of p27 is an early pathogenic event in type I endometrial cancer (ECA).Citation1,49,50 In ECA cell lines, shown to express TGF-β receptors, and primary normal endometrial epithelial cells (EECs), TGF-β signaling through Smad2 and Smad3 inhibited cell proliferation by preventing ubiquitin mediated degradation of nuclear p27 to block Cdk2 activity, with no effect on transcription.Citation49 In contrast, primary ECA cells have aberrant TGF-β signaling leading to perpetual degradation of p27 and loss of TGF-β-mediated growth inhibition.Citation1,2 Knocking-down p27 completely obviated TGF-β mediated growth inhibition implicating p27 as the key cell cycle protein for growth arrest by this cytokine.Citation49 Further, these studies showed that TGF-β protects p27 from proteasomal degradation in these cell lines by decreasing Skp2 and Cks1 E3 ligase activity. Consistent with the mitogenic effects of estrogen on the endometrium, Type I endometrial cancer (85% of endometrial cancers) is induced by unopposed estrogen.Citation51 Estrogen causes both MAPK/ERK2-dependent phosphorylation of p27 on Thr187 required for its identification and ubiquitylation by Skp2/Cks1 and accordingly, Skp2 deletion studies showed that the estrogenic effects on cell proliferation due to p27 degradation are Skp2-dependent.Citation52 Conversely, progesterone inhibits estrogen induced proliferation of the endometrium in the secretory phase of the menstrual cycle and accordingly, progesterone a therapeutic for endometrial hyperplasia and grade I carcinoma,Citation53 markedly increases the levels of nuclear p27 and decreases Skp2 and Cks1.Citation52 The opposing effects of estrogen and progesterone are recapitulated through their inverse effects on the levels of Cdh1 and its E3 ligase activity for Skp2 and Cks1 when bound to APC; specifically, estrogen decreases and progesterone increases Cdh1.Citation52 Knocking down Cdh1 completely obviates progesterone-induced increase in nuclear p27 and progesterone-mediated growth inhibition. Thus, Cdh1 is the upstream controlling factor in the regulation of endometrial growth by hormones, which converge on p27 cell cycle regulation. In addition, we showed that TGF-β decreases Skp2 and Cks1 in HEC-1A cells, which was blocked by the proteasome inhibitor, lactacystin thereby implicating the E3 ligase APC/Cdh1 in the destruction of Skp2 and Cks1 in these cells.Citation49,54 In the present study, we show that TGF-β Smad2/3 dependent signaling increases the levels of Cdh1 only in the nucleus where it is found to reside in primary EECs and in ECA cell lines. Moreover, TGF-β activates the APC by inducing Cdh1 binding to the APC to form APC/CCdh1 E3 ligase complex. Consistent with the tumor suppressor role for Cdh1, the abundance of APC/CCdh1 increases the turnover of Cks1 and Skp2 to enable TGF-β-induced inhibition of proliferation through an increase in nuclear p27. Knocking down Cdh1 both, abolished the TGF-β-induced increase in nuclear p27 and stabilized Skp2 and Cks1. Moreover, protein synthesis was not required for TGF-β induced increase in p27 and decreases in Cks1 and Skp2. The effect of TGF-β on Cdh1 transcription appears to be minimal in ECA cell lines and normal primary EECs but there is no effect on Skp2 and Cks1 mRNA levels. Therefore, these data suggest that TGF-β ensures the preservation of nuclear p27 to control proliferation in EECs by exploiting the UPS. The loss of expression of nuclear p27 due to its persistent degradation is associated with the high levels of Skp2 and Cks1 in vitro and in vivo in ECA. Therefore, preventing p27 degradation by increasing Cdh1 is a rational therapeutic approach to regain cytostatic control. Moreover, the TGF-β-induced increase in nuclear Cdh1, that protects p27 from being degraded appears to be a key mechanism underlying dysregulated TGF-β signaling leading to the breakdown in growth regulation observed early in ECA development.
Results
TGF-β prevents p27 ubiquitin-mediated degradation by increasing nuclear Cdh1 in ECA cell lines
We determined whether TGF-β increases Cdh1 in the nucleus of ECA cell lines and primary EECs to effectuate proteasomal destruction of Skp2/Cks1. This would leave p27 intact to accumulate in the nucleus for cell cycle arrest in ECA cell lines having intact TGF-β signaling. Primary ECA cells were not tested as we previously showed that they lack TGF-β receptors, as a lesion in this disease and thus, were not growth inhibited by TGF-β.Citation2 As shown in , TGF-β induces a dose-dependent increase in Cdh1 protein in ECC-1 and HEC-1A cells (not shown) with a peak response of 1.7-fold at 100 pM TGF-β, which was blocked by SD208, an inhibitor of TβRI kinase activity. In addition, TGF-β (100 pM) time-dependently induces Cdh1 protein () in ECC-1 and HEC-1A cells by 1.3 and 1.2-fold, respectively, in 2 h, with a peak response of 1.8-fold at 48 h in ECC-1 cells and 1.9-fold at 72 h in HEC-1A cells. TGF-β induced Cdh1 transcription at 48 h in both cell lines by 4.2-fold and 2.4-fold in ECC-1 and HEC-1A cells, respectively ().
Figure 1. TGF-β dose-dependently and time-dependently increases nuclear Cdh1 while decreasing Skp2 and Cks1 to prevent ubiquitin-mediated degradation of p27 in ECA cell lines. (A) TGF-β dose-dependently induces Cdh1 protein. ECC-1 cells, were treated with increasing concentrations of TGF-β in the presence or absence of 1.0 µM SD208 (an inhibitor of TGFβRI/Alk5 kinase activity) for 48 hours and total cell lysates subjected to immunoblotting (left panel) and densitometry of the bands (right panel). Each band was scanned and normalized to the intensity of β-actin for each sample/well and values determined compared to untreated control set at 100%. The data are expressed as per cent relative intensity. The blot represents one independent experiment of n = 3. (B) TGF-β time-dependently induces Cdh1 protein. ECC-1 cells and HEC1-A cells were treated with 100 pM TGF-β, and cell lysates prepared at the indicated time points. Lysates were subjected to immunoblotting followed by densitometry and relative intensity determined as described in (A). (C) TGF-β effect on Cdh1 mRNA levels. HEC-1A cells were treated with 100 pM TGF-β for increasing times and subjected to quantitative Real-Time PCR. Relative gene expression was calculated against β-actin as internal control using the ΔΔCT method. The graph shows Cdh1 mRNA represented as fold change compared to untreated control (Unt). Error bars represent standard deviation calculated from 3 independent experiments. (D) Effect of TGF-β on Cdh1, p27, Skp2, and Cks1 protein levels in nuclear and cytoplasmic fractions. ECC-1 cells were treated with 100 pM TGF-β in the presence or absence of 1.0 µM SD208 or 1.0 µM lactacystin (Lac) for 48 hours. Following immunoblotting of total cell lysates and subcellular fractions with appropriate antibodies, protein levels were determined by densitometry of the bands and relative intensity calculated as shown in the graph below the blot and as described in (A). (top panel, representative blot from n = 2).
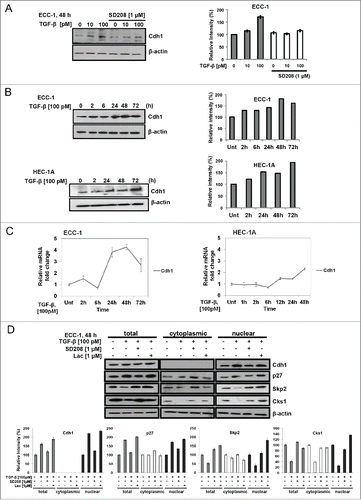
The increase in Cdh1 induced by TGF-β is expected to be nuclear to affect the cell cycle for inhibition of proliferation. Subcellular fractionation of ECC-1 cells following TGF-β treatment showed that the increase of Cdh1 (2.2-fold) occurred exclusively in the nucleus and it is notable that Cdh1 does not reside in the cytoplasm in these cells (). By densitometry, the increase in nuclear Cdh1 was accompanied by a 1.8-fold increase in p27 and a concomitant decrease of Skp2 (61%) and Cks1 (75%). Ensuring that the TGF-β-driven responses were directly due to TGF-β signaling via the downstream transcription factor Smad2 and Smad3, in the presence of SD208, all responses were fully or nearly (i.e., Cks1) reversed to untreated control levels in the nucleus. Importantly, Skp2 and Cks1 nuclear levels were increased over the untreated control following treatment with the proteasome inhibitor lactacystin (Lac; 1.0 μM) confirming that TGF-β decreases Skp2 and Cks1 levels through degradation in proteasomes and furthermore, supporting the role of the increase in Cdh1 by TGF-β in the regulation of Skp2/Cks1 E3 ligase activity. TGF-β also decreases the cytoplasmic level of Skp2 and Cks1 by 18% and 62%, respectively but had no apparent effect on p27. The effect of TGF-β on total and cytoplasmic p27, Skp2 and Cks1 levels was either completely or partially reversed by SD208. Thus, the increase in Cdh1 and p27 shown in the total lysates was due to the increase in the nuclear fraction through TGF-β signaling (inhibited by SD208).
TGF-β increases Cdh1 and decreases Skp2 and Cks1 to stabilize nuclear p27 in primary endometrial epithelial cells
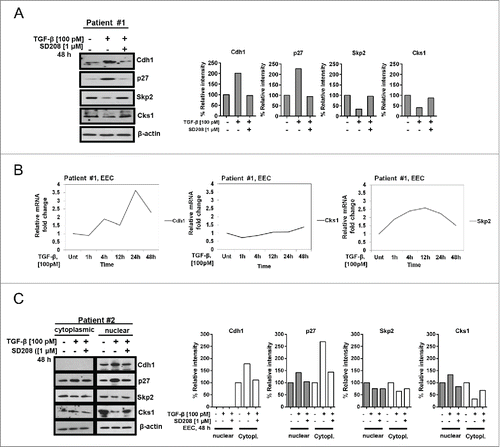
Similar to the results obtained with the ECA cell lines, treatment of primary EECs (normal proliferative phase endometrium [PE]; Patient #1) with 100 pM TGF-β for 48 h induces a 2-fold and 2.3-fold increase in Cdh1 and p27, respectively (). A concomitant decrease of 66% and 59% in Skp2 and Cks1 protein levels, respectively, was shown which, was blocked by SD208. In contrast, in the same Patient #1, TGF-β did not affect mRNA transcription of Skp2, or Cks1 up to 48 h but a small increase in Cdh1 of 3.6- fold was observed at 24 h post-treatment (). Following subcellular fractionation of normal EECs from secretory phase endometrium (SE; Patient #2) treated with TGF-β for 48 h in vitro, Cdh1 and p27 vary directly with 1.8 and 2.1-fold increases in the nucleus, respectively. As expected, the basal level of p27 in primary cultures of normal proliferating endometrium (PE) is negligible whereas basal levels of p27 are found in the nucleus and cytoplasm in SE from Patient #2 (). Inverse to Cdh1 and p27, Skp2 and Cks1 vary directly showing a 31% and 72% decrease, respectively (). Each respective protein response was completely blocked by SD208, again indicating that the changes in protein levels are mediated through TGF-β signaling. Interestingly, as observed for ECC-1 cells, Cdh1 is not present in the cytoplasm in normal EECs (SE) shown here and the effects of TGF-β on the cytoplasmic levels of the Skp2/Cks1-p27 axis are unremarkable.
Figure 2. TGF-β increases Cdh1 and decreases Skp2 and Cks1 to stabilize nuclear p27 in primary endometrial epithelial cells (EECs). (A) TGF-β effect on the protein levels of Cdh1, p27, Skp2, and Cks1. Normal primary EECs isolated from proliferative phase endometrium (Patient #1) were synchronized for 24 h, treated with 100 pM TGF-β under serum-free conditions for 48 h in the presence or absence of SD208 (1µM), and total cell lysates subjected to immunoblotting (left panel) followed by densitometry (right panel). Each band was scanned and normalized to the intensity of β-actin for each sample/well and values determined compared to untreated control set at 100%; the data are expressed as per cent relative intensity. The blot represents one independent experiment of n = 3. (B) TGF-β effect on Cdh1, Skp2, and Cks1 mRNA levels. Normal primary EECs from Patient #1 were synchronized, as described in (A), treated with 100 pM TGF-β for increasing times, as indicated, and subjected to quantitative Real-Time PCR. Relative gene expression was calculated against β-actin as internal control using the ΔΔCT method. The graph represents relative fold-change in mRNA levels compared to the untreated sample (Unt). (C) Effect of TGF-β on Cdh1, p27, Skp2, and Cks1 protein levels in nuclear and cytoplasmic fractions. Normal primary EECs were isolated from secretory phase endometrium (Patient #2) and the cells treated with 100 pM TGF-β, as described above in (A). Subcellular fractions were collected and protein levels determined by immunoblotting (left panel) and densitometry (right panel). Per cent relative intensity was calculated as described above in (A).
The increase in TGF-β-induced Cdh1 is associated with TGF-β-mediated cell cycle arrest and increased binding of Cdh1 to the Anaphase Promoting Complex (APC) in ECA cell lines
Whereas TGF-β signaling utilizes different CDKIs for its growth inhibitory response in different epithelial cell types, the CDKI, p27 is the sole driver of this response in endometrial normal and malignant cells (expressing TGF-β receptors).Citation49 shows that TGF-β dose-dependently inhibited proliferation of ECC-1 cells with a peak response of 37% at 100 pM at 48 h. Inhibition of proliferation was completely reversed by SD208 thereby implicating that TGF-β signaling is intact and indeed, TβRI and TβRII are present on these cells (). Thus, classic Smad2/3 downstream signaling mediates this response. Consistent with TGF-β mediated inhibition of proliferation, TGF-β (100 pM) increases the number of ECC-1 cells in G1 from 53% to 61% while decreasing the number of cells in S phase from 29% to 22% at 48 h (). This modest transition of cells into G1 was observed with progesterone treatment of ECC-1 cells for 48 h and likely reflects the modest inhibition of proliferation of these synchronized cells in monolayer cultures.Citation52 Following treatment of ECC-1 cells on chamber slides with 100 pM TGF-β for 48 h, as shown by the graph (right) a 6.7-fold increase in Cdh1 nuclear immunostaining (, right panel) is observed compared to the untreated controls (, left panel; 47% of cell nuclei stained compared to 7%; p≤0.0001). Moreover, as shown by immunoblotting (), Cdh1 is exclusively localized to the nucleus. In addition, similar to growth inhibition determined by the MTS assay (; 37%), there is 34% decrease in the number of cells on the slides following TGF-β treatment.
Figure 3. TGF-β-induces inhibition of proliferation concomitant with an increase in Cdh1 and binding of Cdh1 to the Anaphase Promoting Complex (APC) in ECA cell lines. (A) TGF-β inhibits cellular proliferation. ECC-1 cells were synchronized, treated with increasing concentrations of TGF-β1 (0–100 pM) for 48 h in the presence or absence of SD208, and cell proliferation assessed by the MTS assay. Inhibition of proliferation (growth) is expressed as per cent of the untreated control at 100%. Error bars represent standard error from the mean of 3 independent experiments, **p ≤ 0.01, ****p ≤ 0.0001. As shown by the immunoblot (inset), ECC-1 cells express TGF-β receptors, TβRI and Tβ RII. (B) TGF-β increases the percent of cells in G1. ECC-1 cells were synchronized, treated with 100 pM TGF-β or untreated for 48 h, and cell cycle analysis performed using propidium iodide staining. The graph represents the per cent of cells in each phase of the cell cycle in TGF-β treated cells compared to untreated controls (n = 2). (C) TGF-β increases the number of cells expressing Cdh1. ECC-1 cells were plated on chamber slides, synchronized, treated with 100 pM TGF-β or left untreated for 48 h, and subjected to immunocytochemistry using anti-Cdh1. The brown stained nuclei (left side, right panel) designates cells expressing Cdh1. The graph (right side) represents the average of the number of Cdh1-negative (Cdh1-) and Cdh1 positive (Cdh1+) cells per 4 fields in untreated and TGF-β- treated cells. The bars indicate the mean ± SD. Expression of Cdh1 in TGF-β-treated versus untreated cells (****p ≤ 0.0001) and decrease in the total number of cells (p < 0.089; n = 3). (D) TGF-β induces Cdh1 binding to APC for activation of Cdh1/APC E3 ligase. ECC-1 cells were synchronized, treated with TGF-β (100 pM) or untreated (Unt) for 48 h, cell lysates immunoprecipitated with anti-Cdh1, and subsequently immunoblotted with anti-Cdc27/Apc3. The blot represents one independent experiment of n = 4.
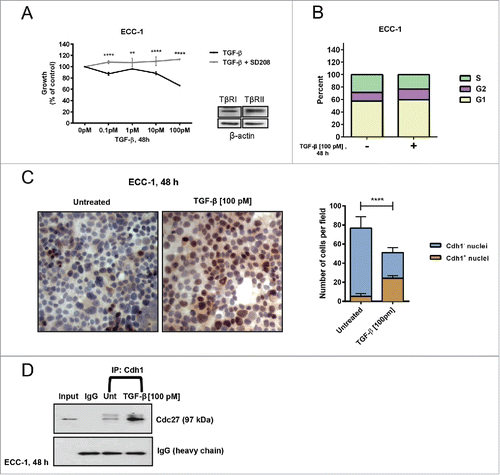
The requisite E3 ligase ubiquitlylating activity for Skp2 and Cks1 substrates requires the binding of Cdh1 to the APC in a complex through the displacement of Cdc20 at G1 phase of the cell cycle (enabled by Cdc14 phosphatase activity on Cdh1). To determine whether the TGF-β-driven increase in Cdh1 concurrently induces the binding (activation) of Cdh1 to the APC, ECC-1 cells were treated with 100 pM TGF-β, immunoprecipitated with anti-Cdh1, and immunoblotted with anti-Cdc27/Apc3, an externally located core component of the APC/C that also binds Cdh1.Citation19 Indeed we show in , that TGF-β increased the association between Cdh1 and the APC complex suggesting that in addition to the increase in [nuclear] Cdh1 observed by immunoblotting and immunocytochemistry, TGF-β also increases APC/CCdh1 E3 ligase activity for proteasomal degradation of Skp2 and Cks1 leaving p27 intact in the nucleus to function in cell cycle arrest.
Cdh1 is required for TGF-β-mediated inhibition of proliferation and stabilization of nuclear p27
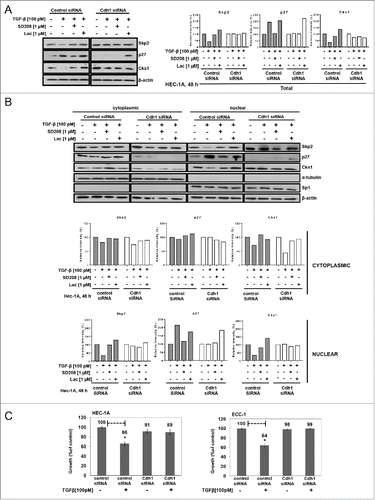
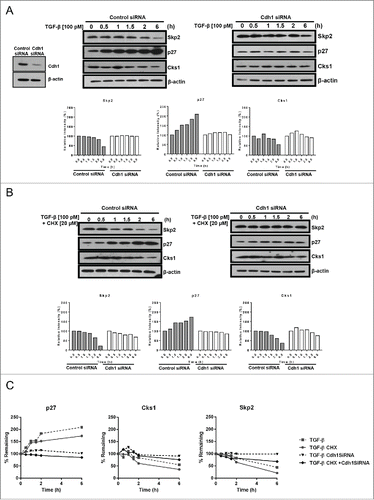
We show herein that TGF-β increases Cdh1 and p27 while inversely affecting the levels of Skp2 and Cks1 in the nucleus [through APC/CCdh1 E3 ligase activity] concomitant with cell cycle arrest. Therefore, to determine whether Cdh1 is required for TGF-β-mediated inhibition of proliferation through stabilization of nuclear p27, we transiently depleted Cdh1 in ECC-1 cells using Cdh1 (fzr) siRNA and achieved 82% reduction in Cdh1 in the nucleus (Cdh1 is not found in the cytoplasm in ECA cell lines) compared to cells transfected with control siRNA (Figure S1). As shown in , of total cell lysates and sub-fractionated lysates, respectively, TGF-β (100 pM) increases total and nuclear p27 by 1.9-fold and 2.1-fold, respectively, and inversely, decreases total Skp2 and Cks1 by 58% and 66%, respectively, in the control siRNA transfected cells (n.b., results are similar to those of non-transfected ECC-1 cells shown in ). However, following Cdh1 knock-down and thus, subsequent loss of E3 ligase activity for Skp2 and Cks1, expectedly, the basal levels of Skp2 and Cks1were markedly increased compared to the control siRNA treated cells (). In addition, the TGF-β-induced decrease in Skp2 and Cks1 and increase in p27 was abolished as shown in both the total cell lysates and nuclear fractions. Notably, lactacystin did not recover nuclear or cytoplasmic levels of p27. TGF-β only slightly decreased Skp2 and Cks1 and had no effect on p27 in the cytoplasm (). The presence of SD208 blocked all TGF-β-mediated responses again, ostensibly ensuring TβRI/RII/Smad2/3 signaling causes the observed effects. Functionally, as shown in , knocking-down Cdh1 abrogated TGF-β-mediated inhibition of proliferation compared to control siRNA transfected cells, which were inhibited by 34% and 36% in HEC-1A and ECC-1 cells, respectively; similar results to those shown in . These results demonstrate that TGF-β-mediated inhibition of proliferation in ECA cells occurs through induction of Cdh1 protein.
Figure 4. Cdh1 is required for TGF-β-mediated inhibition of proliferation and stabilization of nuclear p27. (A) Knocking-down Cdh1 blocks TGF-β induction of p27 and increases Skp2 and Cks1. HEC-1A cells were transiently transfected with Cdh1 siRNA or control siRNA, incubated overnight in media containing 10% FBS, synchronized, and treated with 100 pM TGF-β in the presence or absence of 1.0 µM SD208 or 1.0 µM lactacystin (Lac) for 48 h. The expression of Skp2, p27, and Cks1 in total cell lysates was quantified by immunoblotting (left panel) and densitometry (right panel). Each band was scanned and normalized to the intensity of β-actin, as a loading control, for each sample/well and values determined compared to untreated control set at 100%. The data are expressed as per cent relative intensity. The blot represents one independent experiment of n = 3. (B) Knocking-down Cdh1 blocks TGF-β induction of p27 and increases Skp2 and Cks1 in nuclear fractions. Following transfection of Cdh1 siRNA and treatments as described in (A), the cells were subjected to subcellular fractionation and protein levels of Skp2, p27, and Cks1 protein levels quantified by immunoblotting (upper panel) and densitometry, (lower panel), as described in (A). The blot represents one independent experiment of n = 3. (C) Knocking down Cdh1 blocks TGF-β-mediated inhibition of proliferation. HEC-1A and ECC-1 cells were transfected with Cdh1 siRNA or control siRNA and treated or untreated with 100 pM TGF-β for 48 h, as described in (A); cell proliferation was assessed by the MTS assay. Inhibition of proliferation (growth) is expressed as per cent of untreated control set at 100%. The data are expressed as mean ± SEM from 3 independent experiments of quadruplicate samples, p ≤ *0.05.
The TGF-β-mediated changes in the levels of p27, Skp2, and Cks1 do not require protein synthesis in Cdh1 replete cells and Skp2 and Cks1 half-life is extended in Cdh1-deficient cells
To quantitate the effect of TGF-β on the protein turn-over of Skp2 and Cks1 through its regulation of Cdh1, ECC-1 cells expressing nuclear Cdh1 (control siRNA-treated) or with Cdh1 transiently knocked-down with siRNA were treated with TGF-β (100 pM) in the presence or absence of cycloheximide (CHX; 20 µM); knock-down efficiency was 89% (, left panel, inset). As shown in (left panel), in the Cdh1 replete control cells, TGF-β progressively decreases the levels of Skp2 and Cks1 over time with a 56% and 45% decrease, respectively, at the final time point of 6 h. Concomitantly, TGF-β progressively increases p27 culminating in the consistently observed 2-fold increase at 6 h. By contrast, Skp2, Cks1, and p27 protein levels were nearly unaffected by TGF-β through-out the time course in the Cdh1-deficient cells (; right panel). With protein synthesis inhibited by CHX, TGFβ-treatment of control siRNA transfected ECC-1 results in a 35% and 79% decrease of Skp2 and a 39% and 64% decrease in Cks1, after 2 and 6 h, respectively (, left panel). Consistent with the TGF-β-induced decrease in Skp2 and Cks1, the level of p27 protein was increased by 1.9-fold at 6 h (, left panel). In sharp contrast to TGF-β effects on Skp2, Cks1 and p27 in the presence of Cdh1 (wild-type), in the Cdh1-depleted cells (; right panel), the TGF-β-induced effects on Skp2, Cks1 and p27 were attenuated in the presence of CHX (protein remaining: Skp2, 79% at 2 h, 68% at 6 h; Cks1, 93% at 2 h, 76% at 6 h compared to 19% for Skp2 and 30% for Cks1 at 6 h in Cdh1 competent cells (, left panel). In addition, instead of the 2-fold increase in p27 induced by TGF-β in Cdh1 positive cells, in the absence of Cdh1, the increase in p27 levels was blocked and interestingly, although there was ample Skp2 and Cks1 shown in these total cell lysates (, right panel) to ostensibly cause degradation of p27, p27 was not decreased but essentially remained unchanged with and without CHX (, right panel). The line graphs shown in demonstrate the changes in the Skp2, Cks1, and p27 proteins in the Cdh1 wild-type and Cdh1-deficient cells following treatment with TGF-β or, with TGF-β in the presence of CHX over time. Notably, the half-life of both Cks1 and Skp2 was extended with or without CHX in the Cdh1-depeleted cells treated with TGF-β and thus, protein synthesis was not involved in the consistent preservation of Cks1 and Skp2 protein levels within 6 h. Taken together, expressed as per cent remaining proteins over time up to 6 h, we estimate that the half-life of Skp2 and Cks1 is increased to 9.4 and 9.7 h, respectively, in the Cdh1-deficient cells compared to 3.85 and 4.2 h, respectively, in the Cdh1 competent cells. Hence, in the absence of Cdh1, TGF-β is unable to protect p27 from degradation (reflected by no accumulation of a p27 over 6 h) and both Skp2 and Cks1 proteins are stabilized with increased half-lives of 5.6 and 5.5 h, respectively.
Figure 5. The TGF-β-mediated increase in p27 and decrease in Skp2 and Cks1 do not require protein synthesis in Cdh1 wild type cells; TGF-β-induced effects are obviated in Cdh1-deficient cells and Skp2 and Cks1 half-lives are extended. (A) TGF-β effects on p27, Skp2, and Cks1 protein levels in Cdh1-deficient cells (right panel) compared to siRNA control cells (left panel). ECC-1 cells were synchronized, treated with 100 pM TGF-β, and cell lysates prepared at the times indicated. The lysates were subjected to immunoblot analysis followed by densitometry. Cdh1 knockdown efficiency of 89% is shown by western blot analysis (inset left). Each band was scanned and normalized to the intensity of β-actin as a loading control for each sample/well and values determined compared to untreated control set at 100%, as shown in the graphs (bottom panels). The data are expressed as per cent relative intensity. The blot represents one independent experiment of n = 3. (B) TGF-β effects on p27, Skp2 and Cks1 protein synthesis in Cdh1-deficient cells (right panel) and siRNA control cells (left panel) in the presence or absence of cyclohexmide (CHX). ECC-1 cells were treated as described in (A) with 100 pM TGF-β in the presence or absence of 20 µM CHX for the times indicated. Cell lysates were prepared and analyzed as described in (A). One representative blot from n = 3. (C) TGFβ-mediated increase in Cdh1 stabilizes nuclear p27 and decreases Skp2 and Cks1 half-lives in Cdh1 siRNA control cells (wild type Cdh1) cells. Skp2 and Cks1 half-lives are extended in Cdh1 deficient cells. The graph depicts per cent protein remaining over time (6 h) in TGF-β (100 pM)-treated control siRNA or Cdh1 siRNA- treated cells in the presence or absence of CHX derived from densitometry of the blots in panels A and B; average of n = 3 experiments.
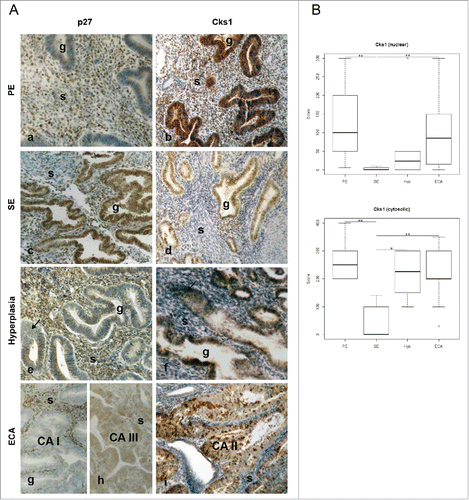
p27 and Cks1 are inversely expressed in normal endometrium and all grades of type I endometrial cancer in vivo recapitulating their growth regulatory effects in vitro
As we show that Cks1 protein levels vary directly with Skp2, in vitro, and Cks1 is rate limiting to the E3 ligase activity of the SCF-Skp2/Cks1, to determine whether Cks1 levels similarly vary with Skp2 in vivo, we performed immunohistochemical analysis for the presence of Cks1 in the nucleus and cytoplasm of normal glandular epithelium of proliferative phase (PE), secretory phase (SE) endometrium, simple and complex hyperplasia (HYP), and in increasing grades of type I ECA. The expression of Skp2 was previously shown to be increased in ECA patients compared to normal tissue.Citation55 As shown in , there is an inverse relationship between p27 (left panel) and Cks1 (right panel) immunoreactivity in both normal (Panels a-d), hyperplastic (Panels e-f) and malignant tissue (Panels g-i). The level of expression of p27 and Cks1 in the glandular epithelium (g) of both PE and SE is consistent with the role of p27 in cell cycle progression and arrest, respectively. PE glands show very slight p27 nuclear staining (Panel a) whereas immunostaining is highly intense for Cks1 in the nucleus in these cells (Panel b); a large subpopulation of stromal (s) cells express both p27 and Cks1. In sharp contrast, there is intense nuclear expression of p27 in the differentiated glandular epithelial cells of SE and in a subpopulation of stromal cells (Panel c) whereas there are a small number of cells within the glands that express nuclear and cytoplasmic Cks1 in these ostensibly non-dividing cells (Panel d). Moreover, less stromal cells are stained for Cks1 in SE compared to PE (compare Panels b and d). In pre-neoplastic hyperplasia, the glands show evidence of hyperproliferation (arrows) and a loss of nuclear p27 with slight cytoplasmic immunoreactivity (Panel e; simple hyperplasia). In contrast, Cks1 immunoreactivity is intense in most of the glandular epithelial cells (Panel f). With increasing grade of ECA characterized by increased proliferation of glandular epithelial cells causing glandular crowding, the stromal cells surrounding the glands are incrementally reduced in number, as shown in Panels g-i. As expected from the data herein showing the inverse relationship between p27 and Cks1 in the ECA cell lines and primary ECA cells by western blotting (), Panel g (grade I ECA) and Panel h (grade II ECA) show near absence of p27 expression in the malignant glands. In contrast, Panel i (grade II ECA) shows punctate nuclear immunoreactivity for Cks1 with no staining of remaining stromal cells. A role for Cks1 in endometrial stromal cells has not been documented. However, the stromal cell immunostaining for Cks1 appears to be higher in normal PE and lower in SE and cancer tissues implicating a differential function of Cks1 in these cells. No immunoreactivity was observed using non-immune rabbit serum as a negative control (not shown). The box plots in show a comparison of the scores for nuclear and cytoplasmic Cks1 immunostaining in PE (n = 7), SE (n = 5), hyperplasia (Hyp; composed of simple, complex without atypia, and complex with atypia histology; Hyp, n = 10), and grades I-III ECA (n = 22). Subjected to the statistical analyses, the scores among increasing grades of ECA were not statistically different and thus, were grouped together. Comparing nuclear values of intensity of immunostaining for Cks1 across PE, SE, Hyp and ECA, the Kruskal-Wallis test shows a significant difference in values (Chi-Square = 13.30; p = 0.004). A Dunn's Multiple Comparison Test shows statistically significant differences between SE compared to PE and SE compared to ECA (p < 0.01). Comparing cytoplasmic values across categories, the Kruskal-Wallis test shows significant difference (Chi-Square-13.11; p = 0.0044). The values for ECA, Hyp, and PE are each significantly different compared to SE (PE vs SE, P < 0.002); Hyp vs SE, p < 0.013; ECA vs SE, p < 0.005).
Figure 6. Immunohistochemical analysis of normal, hyperplastic, and type I endometrial (endometrioid) cancer tissues shows inverse expression of p27 and Cks1. (A) Tissues from normal proliferative (PE; panels a-b) and secretory (SE; c-d) endometrium, endometrial hyperplasia panels (HYP; e-f), and ECA (panels g-i) were immunostained for Cks1 or p27. The samples were scored as described in Materials and Methods. (B) The box plot represents nuclear and cytosolic immunoreactivity scores for Cks1 in normal PE (n = 7) and SE (n = 5), HYP (n = 10), and ECA (n = 22). Immunostaining scores for p27 were previously reported.Citation1 The Kruskal-Wallis nonparametric tests were performed to assess the difference in Cks1 values (nuclear and cytosolic) across disease category followed by Dunn's Test for multiple comparisons (* P ≤ 0.05, ** P ≤ 0.01. g = Gland; s = stromal cells). Magnification for all is 10×. To improve visual matches of photos of slides, panel d was adjusted to remove excessive pink color.
Discussion
We previously showed that TGF-β does not inhibit proliferation of primary ECA cells (isolated from hysterectomies) due to disabled TGF-β signaling but that primary normal EECs isolated from PE are growth inhibited by 55% with 10 pM TGF-β; differentiated normal SE cells are less sensitive to TGF-β.Citation2,56,57 Interestingly, we show that ECA immortalized cell lines express TGF-β receptors that function to mediate inhibition of proliferation. Once TGF-β can no longer signal for growth inhibition early in neoplastic progression, TGF-β receptor canonical signaling pathway can mediate oncogenic functions of TGF-β.Citation5,58 Therefore, TGF-β receptor expression can be mutable with respect to culture of immortalized cell lines compared to primary ECA cells. As loss of nuclear p27 is observed in endometrial hyperplasia, the precursor to type I ECA, which is linked to unopposed estrogen stimulation,Citation51 we concluded from our studies that estrogen-induction of p27 ubiquitin-mediated degradation and/or dysregulated TGF-β signaling in ECA are early events involved in the pathogenesis of type I endometrial cancer.Citation1,2,49,52 As we previously showed that TGF-β treatment of ECA cell lines decreases Skp2 and Cks1, the aim of the current studies was to determine whether TGF-β induces APC/CCdh1 E3 ligase activity to cause degradation of Skp2 and Cks1 as a means for maintaining sufficient levels of nuclear p27 in ECA cell lines and primary EECs to inhibit cell growth. Similar to progesterone induction of Cdh1 shown previously,Citation52 TGF-β dose and time-dependently induces nuclear Cdh1. TGF-β has also been shown to induce translocation of Skp2 from the cytoplasm to the nucleus to facilitate Skp2 ubiquitylation by APC/CCdh1 complex.Citation59 The role of Cdh1 as a tumor suppressor was shown in a xenograft model of human breast cancer growth.Citation60 Overexpression of Cdh1 resulted in decreased tumor growth and conversely, depletion of Cdh1 enhanced tumor growth. Furthermore, as we have shown for ECA cell lines and primary human EECs, these studies showed an inverse relationship between Skp2 and Cdh1 and that Cdh1 varied directly with p27 in the normal breast cell line, MCF10A, and breast cancer cell lines. Low levels of Cdh1 were demonstrated in cancer tissue compared to normal counterparts in colon, rectal and breast cancer but not in other cancers such as prostate and brain and, in some cancers low Cdh1 has been correlated with high levels of Skp2.Citation61
Whereas previous studies by others focused on the effect of TGF-β on Skp2 degradation via APC/Cdh1 activity,Citation21 we show here that TGF-β also induces a concomitant decrease in the Skp2 adaptor protein, Cks1, in the nucleus and cytoplasm in ECA cell lines and EECs. This is consistent with Cks1 as the rate limiting component of Skp2/Cks1 E3 ligase activity that enables p27 ubiquitin-mediated degradation.Citation37 In another study, using an in vitro ubiquitylation assay with purified recombinant proteins and TGF-β-treated mink lung carcinoma cell line (Mv1Lu) extracts, the addition of recombinant Cks1 was necessary for TGF-β-driven Skp2 ubiquitylation of p27.Citation36 However, the effects of TGF-β on Cks1 levels, as they vary with respect to the APC/Cdh1-Skp2/Cks1- p27 axis, were not investigated. Whereas TGF-β was shown to decrease Cks1 transcription (5 h) in Mv1Lu cells, we did not observe this effect in the ECA cell linesCitation49 or primary EECs. Taken together, our data implicate Cdh1 as the master upstream regulator of p27 stabilization to regain growth control and therefore, as a significant therapeutic target for ECA. In fact, small molecule inhibitors of Skp2/Cks1 have recently been shown to block estrogen-induced endometrial hyperplasia in mice with an increase in nuclear p27 and strong inhibition of proliferation of glandular endometrial cellsCitation62 again, emphasizing the lack of nuclear p27 as a significant therapeutic target in endometrial cancer and also, in other human cancers that express high Skp2 levels concurrent with lack of nuclear p27. Notably, these inversely related markers are associated with poor survival.Citation63,64
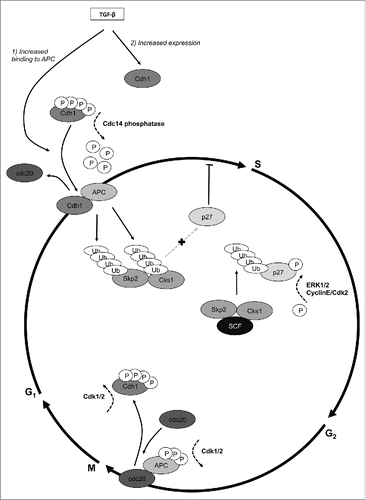
Consistent with the important function of Cdh1 in G1 arrest, we show that basal levels of Cdh1 are present only in the nucleus in ECA cell lines and EECs. Similarly, nuclear sub-localization of Cdh1 has been shown in breast cancer cells and in Mv1Lu cells.Citation59 Possibly, this observation is due to rapid nuclear translocation of Cdh1 following translation on ribosomes enabled by phosphorylation of specific amino acids.Citation65 Interestingly, we show that Cdh1 is mis-localized to the cytoplasm in primary ECA cells and induced to translocate to the nucleus by progesterone, potentially as part of its therapeutic effect marked by increasing p27 in endometrial hyperplasia and lower grade ECAs.Citation52,66 Accordingly, both immunocytochemical () and western blot analysis (, ) show that Cdh1 is increased in the nucleus following TGF-β treatment with a similar extent of growth inhibition (). As illustrated in , TGF-β has a dual effect on Cdh1 as it increases the level of Cdh1 and induced the binding of Cdh1 to APC/C to form the active E3 ligase complex required for Skp2 and Cks1 ubiquitylation. Further shown in , Cdh1 is required to be in a hypophosphorylated state to be bound to the APC/C, which is achieved by the phosphatase Cdc14 causing Cdh1 to displace Cdc20 from APC/C.Citation23,30,31,67 Conversely, hyperphosphorylation of Cdh1 by Cdk1 releases it from the APC thereby causing Cdc20 to be the binding partner with APC/C and having different E3 ligase activity for Cyclins and Securins to ensure exit from mitosis.Citation19,22,68
Figure 7. Regulation of E3 ligase activity of the Anaphase Promoting Complex/Cyclosome (APC/C) by TGF-β. The SCF-Skp2/Cks1 and APC/C are E3 ligases that temporally ubiquitylate cell cycle proteins to regulate the cell cycle. Citation18,19 TGF-β has a dual effect on the APC/C catalytic co-activator, Cdh1: 1). increases Cdh1 at the protein level and 2). increases the binding of Cdh1 to APC/C thereby forming and activating the APC/CCdh1 E3 ligase. The APC/CCdh1 E3 ligase has substrate specificity for Skp2 and Cks1 causing their ubiquitylation and subsequent proteasomal degradation, which occurs in early G1 phase of the cell cycle.Citation19 The degradation of Skp2 and Cks1 by APC/CCdh1 leaves p27 intact to accumulate in the nucleus for cell cycle arrest late in G1. In turn, the SCF-Skp2/Cks1 E3 ligase identifies and ubiquitylates p27 phosphorylated on T187 by either CyclinE/Cdk2 or MAPK/ERK2, causing p27 degradation in late G1/S for cell cycle progression. The specificity of APC/C E3 ligase activity is dependent on, and regulated by, its binding to the catalytic co-activators, Cdh1 and Cdc20 through phosphorylation and ubiquitylation events.Citation19 Cdh1 binds to APC/C through its N-terminal domain, which has 4 critical phosphorylation sites to mediate this interaction.Citation30 In the hypophosphorylated state, which is maintained by Cdc14 phosphatase, Cdh1 remains bound to APC/Cdh1 (late G1). In late G2/early M, Cdh1 is phosphorylated on these same sites by Cdk2 and/or Cdk1 causing Cdc20 to displace Cdh1 on the APC forming the activated E3 ligase APC/CCdc.Citation20 This complex, with substrate specificity for securins and cyclins and other proteins involved in cell cycle progression remains active through the end of mitosis. The cycle continues with Cdh1 displacing Cdc20 in very late M/early G1. The intricate regulation of Cdh1 levels and binding to APC/C by TGF-β conforms to temporal waves of cell cycle E3 ligase activity to effectuate inhibition of proliferation as shown here in endometrial epithelial cells and likely other cell types.
The presence of lactacystin and TGF-β together did not have an additive effect on increasing Cdh1 or p27 levels in the nucleus () suggesting that TGF-β may, at least in part, act via the same mechanism to regulate Cdh1 levels via preventing its degradation and to lesser extent by indirectly regulating transcription (). Also, TGF-β decreases Skp2 and Cks1 levels in the nucleus but cytoplasmic Skp2 was not affected (). This finding is consistent with the lack of APC/CCdh1 in the cytoplasm. In contrast, cytoplasmic Cks1 was attenuated by TGF-β. Different mechanisms likely control both Cks1 and Skp2 levels as it is known that Cdh1 and Skp2 succumb to autodegradationCitation14,69 and the extensive cell cycle protein network that prevents this phenomenon, such as Cks1 protecting Skp2 from destroying itselfCitation14 and Cks1 competing with p27 for binding to Cdk2,Citation70 to name a few. Nonetheless, Cks1 levels might be also regulated, in part, by degradation in the cytoplasm since lactacystin blocks the observed cytoplasmic degradation ().
The TGF-β-induced decrease in nuclear Skp2 and Cks1 and inhibition of proliferation was completely obviated when Cdh1 was knocked down in both ECC-1 and HEC-1A cells () suggesting that Cdh1is necessary and sufficient for the observed decrease in the SCF-Skp2/Cks1 complex to allow accumulation of nuclear p27. Moreover, the increase in Skp2 and Cks1 levels in the nucleus following Cdh1 knock-down did not occur in the cytoplasm. Again, these results are consistent with the presence of Cdh1 only in the nucleus where destruction of Skp2/Cks1 [decreased levels] is observed in ECA cell lines and primary EECs. Importantly, knocking-down Cdh1 profoundly affected the level of p27, which was obliterated in both the nucleus and cytoplasm. However, the p27 level was recovered in the Cdh1 knockdown cells by blocking Skp2/Cks1 E3 ligase activity with lactacystin in the presence of TGF-β. The fact that TGF-β and lactacystin did not increase Skp2 or Cks1 levels over control in the Cdh1-deficient cells, suggests APC/CCdh1 as the only E3 ligase regulating the levels of these proteins in ECC-1 cells. In the presence of CHX, the TGF-β-induced decrease in Skp2 and Cks1 caused a 1.9-fold increase in p27 protein levels at 6 h. The identical increase in p27 ( [left]) mediated by TGF-β in the absence and presence of CHX strongly suggests that ubiquitin-mediated proteolysis and not transcription/translation regulates p27 levels in ECC-1 cells [via APC/CCdh1]. For this same reason, the presence of CHX did not affect the decrease in Skp2 and Cks1 induced by TGF-β thus, as supported by a lack of transcriptional effects, implicates APC/CCdh1 ubiquitin-mediated proteosomal degradation as the major or sole regulator of Skp2 and Cks1 [and thus, p27]. In support of proteasomal regulation of these proteins, it was similarly shown in Mv1Lu cells, that following TGF-β treatment, both p27 and Skp2 levels were nearly identical in the presence or absence of CHX [at 2 h] and Skp2 transcription was not affected.Citation21 Since Skp2 requires Cks1 for its E3 ligase activity for p27, our results show herein that the effects of TGF-β on Cks1 and Skp2 levels are similar in the presence of CHX in both the Cdh1-deficient and wild-type cells thereby supporting their interdependence for the regulation of nuclear p27 levels. The fact that TGF-β induces the same 2-fold increase in p27 at 6 h () and 48 h () incubation times supports the precise incremental, temporal and cyclic nature of cell cycle regulation by the UPS. In the presence of CHX, the half-life of Skp2 in TGF-β-treated Mv1Lu cells was shown to be 60 minutes.Citation21 Comparatively, in ECC-1 cells, following TGF-β treatment, we show that the half-life of Skp2 and Cks1 is approximately 4 h, () and in Cdh1-deficient cells, half-lives are more than doubled to approximately 9.5 h. Interestingly, this might reflect the high sensitivity of Mv1Lu cells to TGF-β-mediated growth inhibition whereas ECA cell lines and EECs are less responsive (70% versus 40% growth inhibition). Therefore, we reason that the faster turnover of Skp2 in Mv1Lu cells in response to TGF-β possibly leads to greater accumulation of p27 for a stronger growth inhibitory effect. After 48 h incubation with or without TGF-β, the presence of Skp2 and Cks1 destroyed p27 in Cdh1-deficient ECC-1 cells (). However, under these same conditions, p27 remained intact in the presence of Skp2 and Cks1 at 6 h incubation ( [right panel]. The lack of Cdh1 might abnormally promote unknown cell cycle effects herein in these studies. Alternatively, the presence of p27 at 6 h reflects the amount of p27 present at this time point compared to 48 h when at least 2 doubling times would have occurred. At this point p27 would have been exposed to Skp2/Cks1 in late G1 causing its ubiquitylation and degradation for entry to S phase.
In addition to Smad2/3 dependent TGF-β signaling that induces APC/CCdh1-mediated destruction of Skp2 and Cks1 for stabilization of p27 for G1 arrest, another TGF-β-Smad3-dependent signaling pathway inhibits proliferation. By this mechanism, Smad3 together with APC/CCdh1, activated by casein kinase II phosphorylation of the APC core protein Cdc27, induces degradation of the transcriptional repressor, SnoN to enable transcription and protein synthesis of the CKIs, p15, p21 to block Cdk4/6 early in G1.Citation28 Moreover, TGF-β decreases Myc transcription via APC, SnoN co-repressor activity, and Smad3. Interestingly, Cks1 is necessary for Myc transcription, which accordingly, was shown to decrease p27 thereby stimulating proliferation.Citation71 Moreover, Myc has also been shown to suppress p27 mRNA. This dynamic and cross-regulation suggests that TGF-β decreases Myc, which in turn obviates the ability of this oncogene to induce Cks1 and also cullin1, a scaffold component of the SCF-Skp2 complex, with a decrease in p27 through feed forward and negative feedback loops.Citation72 In addition, Cks1 has a plethora of protein interactions and activities central to mitosis via Skp2-independent mechanisms that can control the cell cycle through affecting these feedback loops. For example, Cks1 increases Cdc20 transcription,Citation71 which decreases Cdh1 binding to the APC for pro-mitotic effects. Cks1 also binds to, and increases the transcription of Cdk2, interacts with Cdc25 phosphatase, and is considered a master docking agent for normal cell cycle functions.Citation41 Nonetheless, with all the potential complex interactions of TGF-β (e.g., TGF-β induces p27 binding to cyclin E, increases p15 and p21 transcription/translation) on cell cycle proteins and the fact that Cks1 functions independent of TGF-β action, the current studies suggest that TGF-β action in normal and endometrial growth might be strongly or even solely regulated by the UPS.
Expectedly, as its requisite partner in SCF-Skp2/E3 ligase activity, Cks1 expression in ECA in vivo varies directly with Skp2 and is inverse to p27 in normal endometrium and all grades of ECA. Also, concordant with proliferative endometrium, Cks1 is highly expressed in the nucleus (and cytoplasm). Conversely, secretory endometrium shows scant immunoreactivity for Cks1 whereas there is very strong immunostaining for p27 in the nucleus likely to maintain the differentiated state. Grade I and grade II ECA show loss of p27 in the glandular epithelium. Inversely, Cks1 immunostaining is intense in ECA (). Elevated expression of Cks1 as an independent marker for poor prognosis has been observed in human cancers including prostate,Citation73 breast, lymphomas, and multiple myeloma.Citation41 Co-overexpression of Cks1 and Skp2 with inverse relationship to p27 expression has also been reported in breast, oral, colon, and lung (review),Citation41 as we have shown here in ECA.
Our data indicates that normal PE and malignant endometrial growth appear to utilize a similar mechanism of growth regulation seemingly controlled by proteins of the UPS that control the cell cycle. The follicular/proliferative phase of the menstrual cycle is accompanied by incremental increases in mitogenic estrogen, which we showed in vitro causes MAPK-driven phosphorylation of p27 thereby signaling its ubiquitylation by Skp2/Cks1 ().Citation52 This mechanism of targeting p27 for degradation [by estrogen] to normally allow cell cycle progression is likewise operable in ECA as a pathogenic mechanism for the development of estrogen-induced ECA. In contrast, in the normal endometrium, progesterone produced by the corpus luteum in the secretory phase of the menstrual acts as the brake for proliferation by inducing an increase in Cdh1 and therefore, p27,Citation52 thereby preventing uncontrollable growth. Loss of progesterone receptor, genetic, epigenetic, and post-translational aberrancies may all play a role in estrogen-induced type I ECA. However, it is clear that nuclear p27 is a key target to regulate normal and malignant endometrial growth. Therefore, as Cdh1 appears to be the master upstream regulator of p27, it is important to understand how TGF-β and ovarian hormones regulate Cdh1 for the development of preventative and therapeutic approaches for endometrial hyperplasia, the precursor to type I ECA.
Materials and methods
Cell culture and treatments
Type I endometrial carcinoma HEC-1A (ATCC; cat. # HTB-112) and ECC-1 (ATCC; cat. # CRL-2923) cell lines from well-differentiated endometrioid tumors were purchased from American Tissue Culture Collection. HEC-1A cells and ECC-1 cells were incubated at 5% CO2 and cultured in McCoy's 5A (US Biologicals; cat # M2704) and DMEM/F12 (11039-021, Invitrogen), respectively, phenolred-free media supplemented with 10% charcoal-stripped fetal bovine serum (sFBS; BioWest; cat # S162H), 2 mM L-Glutamine (Mediatech; cat# 25-005-CI,) and antimycotic antibiotic solution (Mediatech; 30-004-CI). Primary normal endometrial epithelial cells (EECs; from leiomyomas not involving the endometrial layer) derived from hysterectomies of women ages 35–55 y performed at New York University Langone Medical Center and Bellevue Hospital were prepared and cultured on Primaria® tissue culture plates (BD Biosciences; cat # 353802) in McCoy's 5A media with supplements, exactly as previously described.Citation1,2 The protocol was approved by The Institutional Review Board of New York University (#H9243). For all experiments, the cell lines and primary cells were grown to approximately 70% confluent and synchronized in serum-free media for 24 h prior to adding TGF-β (0, 10 and 100 pM; R&D Systems; cat # 100-B) for the times indicated for individual experiments. In certain experiments, the cells were treated with the proteasome inhibitor Lactacystin (1 μM; Lac); Calbiochem; cat # 426100) or a small molecule inhibitor of TGF-β-Receptor I (TβRI) kinase activity, SD208 (1 μM; Tocris; cat # 3269), which was added 4 h prior to the addition of TGF-β, as previously described.Citation49 To determine p27, Skp2, and Cks1 turn-over in EEC-1 cells without or with Cdh-1 knocked-down, the cells were either untreated or treated with 100 pM TGF-β, chased with the protein synthesis inhibitor Cycloheximide (20 µM; CHX; Sigma-Aldrich; cat# C7698) 6 h prior to the final harvest time, and cell lysates collected at 0.5, 1.0, 1.5, 2 and 6 h time points.
Proliferation MTS assay and cell cycle analysis
ECC-1 cells were seeded onto 96-well plates (BD Biosciences; cat # 351172) at a density of 4 × 103 cells/well, cultured and synchronized as described above, and treated in triplicate with increasing concentrations of TGF-β (0–100 pM) in the absence or presence of SD208, which was added 4 h prior to adding TGF-β. After 48 h, cellular proliferation was assayed using the Cell- Titer96®-Aqueous-One-Solution-Cell-Proliferation Assay (Promega; cat # 63580). Absorbance was measured at 490 nm using a Model 680 Microplate reader (Bio-Rad Laboratories, Hercules, CA). To determine cell cycle distribution by FACS, ECC-1 cells were seeded onto 6 cm tissue culture plates at 6 × 105 cells/plate, grown until 70% confluent, synchronized in serum-free media for 24 h, treated with 100 pM TGF-β for 48 h, and stained with propidium iodide, as described.Citation49 Data is presented as per cent of cells per phase of the cell cycle.
Immunoblotting
Whole-cell lysates were prepared in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl at pH 7.4, 0.25% sodium deoxycholate, 1% NP-40 and 1 mM EDTA) containing 1 mM PMSF, 1 mM DTT, 1 mM NaF, 1 mM Na3VO4, and 1× Protease Inhibitor Cocktail (Sigma-Aldrich; cat # P3840) followed by freeze thawing once and soluble proteins obtained by centrifugation at 14,000 × g for 10 min at 4°C. Where shown, subcellular fractionation was achieved using the NE-PER kit (Thermo Scientific; cat # 78832) according to manufacturer's instructions. All isolation buffers were supplemented with 1x Protease Inhibitor Cocktail, 1 mM NaF, 1 mM Na3VO4, 1mM PMSF, and 1mM DTT. Total protein concentrations for each sample were determined using the Micro BCA Protein Assay Kit (Thermo Scientific; cat # 23235) and equal amounts of protein (20 µg for total lysates and 10 µg for nuclear and cytoplasmic fractions) diluted in Laemmli sample buffer containing 0.1 M DTT, were resolved by SDS-PAGE (12.5%) and electro-transferred onto a nitrocellulose membrane (BioRad; cat #1620112) for one h at 120V. After confirming equal transfer with 0.2% S-Ponceau (Sigma-Aldrich; cat #3504) staining, the membranes were blocked in 5% non-fat dry milk in TBST (20 mM Tris, 0.15 M NaCl, pH 7.4 containing 0.1% Tween20 (Fisher Scientific; cat # BP337) for one h at room temperature followed by incubation with primary antibodies overnight at 4°C. The following primary antibodies, diluted in TBST were used: mouse anti-p27Kip1 (1:1000; Clone 57, Biosciences; cat #BD610242), mouse anti-p45/Skp2 (1:1000; 8D9, Invitrogen; cat #32–3300) rabbit anti-Cks1 (to Cks1 C-terminus; 1:500; Invitrogen; cat #36–6800), mouse anti-Cdh1 (1:1000; DH01; Calbiochem; cat #CC43), rabbit anti-TβRI (1:1000; V-22, Santa Cruz Biotechnology; cat #sc-398), rabbit anti-TβRII (1:1000; C-16, Santa Cruz; cat #sc-220), and mouse anti-Cdc27 (APC-3; 1:500, AF 3.1; Santa Cruz; cat #sc-9972). To verify equal loading of all samples, the blots were stripped, blocked, and re-probed with mouse anti-β-actin (1:12,000; Sigma-Aldrich; A5441). To ensure purity of the cytoplasmic and nuclear fractions, the blots were stripped, blocked and re-probed with mouse anti-α-tubulin (1:10,000; B-5-1-2, Sigma-Aldrich; cat #T5158) and rabbit anti-Sp1 (1:1000; H-225, Santa Cruz; cat #sc14027), respectively. Subsequently, the membranes were incubated with stabilized horseradish peroxidase-conjugated goat anti-mouse (1:2000; 32430, Thermo Scientific) or goat anti- rabbit secondary antibodies (diluted 1:2000; Thermo Scientific; cat #32460) for one h and protein bands detected by chemiluminescence using the SuperSignal West Dura Extended Duration Substrate kit (Thermo Scientific; cat #34075) followed by exposure to HyBlot CL X-ray film (Denville Scientific; cat # E3012). Quantification of the protein bands was determined by densitometric scanning (Office-jet 6110; Hewlett-Packard) of each band and Logic-100 image analysis (Kodak). The intensity of each band analyzed was normalized to the intensity of β-actin for each sample/well and fold increase or % decrease was determined compared to untreated control.
Quantitative Real Time RT-PCR for Cdh1, Skp2, Cks1
Total RNA was isolated from ECC-1 and HEC-1A cells and primary EECs using the RNeasy-Mini-Kit (Qiagen; cat #74104,) and reverse transcription cDNA synthesis was performed using the QuantiTec Reverse Transcription kit (Qiagen; cat#20531). Quantitative PCR reaction mixtures were prepared using iQ SYBR Green mix (Bio-Rad; cat #170–880), 0.5 µl of each forward and reverse oligonucleotide primer set (50 nM each), and 1.25 ng of cDNA template in a total volume of 20 µl. The primers used for SKP2 (Skp2), and CKS1B (Cks1) have been described.Citation49 The primer sequences for Fizzy-related Protein Homolog, FZR1 (Cdh1) are: Forward, FZR1-F: 5′-GTC CAA GCA CGC CAA CGA GCT GGT GA-3′; Reverse, FZR1-R: 5′-GAC ACA GAC TCC TTT GTC GAA CGG-3′. The PCR reactions were performed in parallel with ACTB (β-actin) as a housekeeping gene, using Mx3000P QPCR System (Stratagene) with the following conditions: 95°C for 3 min, followed by a round of 40 cycles for 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. The PCR products were subjected to melt curve analysis to ensure primer specificity. Relative gene expression was calculated against internal control (β-actin) using the ΔΔCT method.
RNA interference for Cdh1
HEC-1A or ECC-1 cells were cultured at 1.5 × 105 cells/well in 12-well plates in media containing 10% FBS. After 24 h, the cells were transfected using HiPerfect transfection reagent (Qiagen; cat #301702) with 10 nM of either FZR1 (Cdh1) siRNA to 3 different FZR1 gene loci including a pool of siRNA (sc-44349, Santa Cruz Biotechnology) or a control siRNA (Santa Cruz; cat #sc-37007). Following 24 h transfection in fresh media containing 10% FBS, the cells were synchronized in serum-free media and then treated with 100 pM TGF-β in the presence or absence of 1 µM SD208, 1 µM Lactacystin for 48 h. For protein turnover experiments, the control siRNA or FZR1 siRNA transfected cells were treated with 20 µM cycloheximide (CHX) to inhibit protein synthesis, for times shown up to 6 h. Cell lysates or subcellular fractionations were analyzed for protein levels as described for immunoblotting. Knock-down efficiency was confirmed by immunoblotting at this same time point at the end of each experiment. Cell proliferation following knock-down was assessed by seeding cells onto a 96-well plate (4 × 103 cells/well), transfecting and then treating the cells with TGF-β for 48 h, and performing the MTS assay, as described above.
Cdh1 binding to APC by immunoprecipitation and immunoblotting
ECC-1 cells, in complete media and seeded on 10 cm-plates (2 × 106 cells/plate), were synchronized in serum-free media for 24 h and treated with TGFβ (100 pM) for 48 h. Whole-cell lysates were prepared as described above for Immunoblotting. The lysates were pre-cleared with 0.75 µg of mouse IgG and 20 µl of Protein A/G PLUS-Agarose beads (Santa Cruz; cat #sc-2003) for 30 minutes. Pre-cleared lysates were incubated with 0.75 mg mouse anti-Cdh1 (DH01) or 0.75 mg mouse IgG overnight and then with 20 µl of Protein A/G PLUS-Agarose beads for 1 h at 4°C; both with end-over rotation. The beads-immunoprecipitate complex was washed 3× in cold RIPA buffer, re-suspended in Laemmli buffer, and equal volumes were subjected to SDS-PAGE (12.5% acrylamide) and immunoblotted with mouse anti-Cdc27/Apc3 (as described above); 10 µg of total lysate (input) and IgG alone served as controls.
Immunocytochemistry for Cdh1
ECC-1 cells were seeded onto 8-well (2 × 104 cells/well) or 4-well (4 × 104 cells/well) polystyrene chamber slides (Miles Scientific, Naperville IL), grown until approximately 70% confluent, synchronized for 24 h in serum-free media, treated with 100 pM TGF-β for 48 h at 37°C, and the experiments terminated by washing the cells with ice cold PBS. The cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) and permeabilized with 0.2% Triton X-100 in PBS for 15 minutes. The slides were blocked with normal goat serum containing 0.5% BSA (Vector Laboratories; cat #SP-5050) for 1 h at room temperature followed by incubation overnight with primary mouse anti-Cdh1 in blocking serum (1:100; DH01) on a rocker at 4°C; non-immune mouse serum served as a negative control. Slides were equilibrated to room temperature, washed in TBS-BSA (0.1%), incubated with biotinylated goat anti-mouse secondary antibody (Vector Laboratories; cat #BA-9200) prepared in blocking solution, for one h at room temperature followed by washing in 0.1% BSA in TBS. Immunoreactivity was determined using the Vectastain Elite ABC kit (Vector Laboratories; cat #PK-6100) prepared in TBS (0.1% BSA) and 0.5% of the substrate, 3,3 diaminobenzidine tetrahydrochliride (DAB;, Sigma-Aldrich; cat #D5637) followed by counterstaining with Hematoxylin Gills's #2 (Fisher Scientific; cat #72504) and mounting the slides with coverslips using Crystal Mount (Sigman-Aldrich; cat #C0612). Unstained and stained cells of untreated and of TGF-β-treated cells were counted in 4 separate fields and average values were obtained. Per cent growth inhibition was determined by adding the difference in the number of cells between treated and untreated plates.
Immunohistochemical analyses for p27 and Cks1
Tissue sections (4 µm) cut from newly embedded and archival paraffin blocks of normal proliferative (PE) and secretory endometrium (SE), hyperplastic endometrium (HYP), and grades I-III endometrioid ECAs were procured from the New York University and Bellevue Tumor Registry and immunostained as described previouslyCitation1 and according to instructions of the Vectastain Elite ABC kit. Briefly, antigen retrieval was performed by gently boiling the slides in Citra buffer (pH 6) in a microwave oven (for p27, 20 min; for Cks1, 10 min). The slides were cooled to room temperature, washed in TBS and non-specific epitopes blocked with normal goat serum in 0.5% BSA for 20 min at room temperature. The slides were incubated overnight at 4°C with the primary antibodies, mouse anti-p27 (0.5 μg/ml; BD Biosciences; 8D9) or rabbit anti-Cks1 (2.5 µg/ml) and subsequently, incubated with the goat anti-mouse (Vector Laboratories; cat #BA-9200) or goat anti-rabbit (Vector Laboratories; cat # BA-1000) secondary antibodies prepared in the appropriate blocking solution for one h at room temperature and washed; normal goat serum was used as a negative control (not shown). The slides were treated with avidin biotin complex (ABC) for 1 h and the brown color reaction was developed using DAB. Slides were counterstained with Gills #2 Hematoxylin and cover slips applied with Permount, all as described.Citation1 The intensity of nuclear and cytoplasmic immunostaining for the glandular/carcinoma cells was assessed by 3 pathologists and scored (immunostaining levels: 0; 1, weak; 2, moderate; 3, intense, 4, highly intense). The values for immunostaining intensity were determined as the product of average staining intensity and the percentage of cells stained at the prevailing intensity.
Statistical analysis
Proliferation data are presented as means ±SD. Using GraphPad Prism software, a 2-tailed Students t test was used to predict statistical significance of the comparison between 2 means. Results were considered significant at *P < 0.05. For immunohistochemical analysis, the Kruskal-Wallis nonparametric tests were performed to assess the difference in Cks1 values (nuclear and cytosolic) across disease category followed by Dunn's Test for multiple comparisons. Cks1 analysis was completed using SAS 9.3 and R statistical software.
Disclosure of potential conflicts of interest
There are no financial interests associated with this manuscript. The authors have nothing to disclose. The authors have complied with all legal requirements pertaining to New York University Medical School-Langone Medical Center.
Supplementary Files
Download MS Power Point (64 KB)Acknowledgments
We greatly appreciate the consistent help of Stella Wong for tissue acquisition and acknowledge Tsivia Hochman, M.S, for her expertise in the statistical analysis of patient tissues analyzed by IHC.
Funding
Supported by R01 CA176502 from the National Cancer Institute, NIH to LIG. Supported by Bridge Funding from NYU School of Medicine to LIG. We acknowledge the Biostatistics CORE of NCI CCSG P30 CA016087.
References
- Lecanda J, Parekh TV, Gama P, Lin K, Liarski V, Uretsky S, Mittal K, Gold LI. Transforming growth factor-beta, estrogen, and progesterone converge on the regulation of p27Kip1 in the normal and malignant endometrium. Cancer Res 2007; 67:1007-18; PMID:17283133; http://dx.doi.org/10.1158/0008-5472.CAN-06-0235
- Parekh TV, Gama P, Wen X, Demopoulos R, Munger JS, Carcangiu ML, Reiss M, Gold LI. Transforming growth factor beta signaling is disabled early in human endometrial carcinogenesis concomitant with loss of growth inhibition. Cancer Res 2002; 62:2778-90; PMID:12019154
- Chen T, Carter D, Garrigue-Antar L, Reiss M. Transforming growth factor beta type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res 1998; 58:4805-10; PMID:9809982
- Gold LI, Lecanda, J. Mechanisms of Cell Cycle Regulation by TGF-b Disabled in Cancer Totowa, NJ: Humana Press, Inc, 2008.
- Massague J. TGFbeta in Cancer. Cell 2008; 134:215-30; PMID:18662538; http://dx.doi.org/10.1016/j.cell.2008.07.001
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11-30; PMID:23335087; http://dx.doi.org/10.3322/caac.21166
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012; 13:616-30; PMID:22992590; http://dx.doi.org/10.1038/nrm3434
- Heldin CH, Moustakas A. Role of Smads in TGFbeta signaling. Cell and tissue research 2012; 347:21-36; PMID:21643690; http://dx.doi.org/10.1007/s00441-011-1190-x
- Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development (Cambridge, England) 2009; 136:3699-714.
- Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev 1995; 9:1831-45; PMID:7649471; http://dx.doi.org/10.1101/gad.9.15.1831
- Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994; 371:257-61; PMID:8078588; http://dx.doi.org/10.1038/371257a0
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18:2699-711; PMID:15545627; http://dx.doi.org/10.1101/gad.1256504
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev 1994; 8:9-22; PMID:8288131; http://dx.doi.org/10.1101/gad.8.1.9
- Wang W, Ungermannova D, Jin J, Harper JW, Liu X. Negative regulation of SCFSkp2 ubiquitin ligase by TGF-beta signaling. Oncogene 2004; 23:1064-75; PMID:14676846; http://dx.doi.org/10.1038/sj.onc.1207204
- Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002; 110:19-32; PMID:12150994; http://dx.doi.org/10.1016/S0092-8674(02)00801-2
- Kim TA, Kang JM, Hyun JS, Lee B, Kim SJ, Yang ES, Hong S, Lee HJ, Fujii M, Niederhuber JE, et al. The Smad7-Skp2 complex orchestrates Myc stability, impacting on the cytostatic effect of TGF-beta. J Cell Sci 2014; 127:411-21; PMID:24259667; http://dx.doi.org/10.1242/jcs.136028
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67:425-79; PMID:9759494; http://dx.doi.org/10.1146/annurev.biochem.67.1.425
- Kurland JF, Tansey WP. Crashing waves of destruction: the cell cycle and APC(Cdh1) regulation of SCF(Skp2). Cancer Cell 2004; 5:305-6; PMID:15093536; http://dx.doi.org/10.1016/S1535-6108(04)00091-1
- Barford D. Structure, function and mechanism of the anaphase promoting complex (APC/C). Quarterly reviews of biophysics 2011; 44:153-90; PMID:21092369; http://dx.doi.org/10.1017/S0033583510000259
- Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008; 112:1415-24; PMID:18260093; http://dx.doi.org/10.1002/cncr.23317
- Liu W, Wu G, Li W, Lobur D, Wan Y. Cdh1-anaphase-promoting complex targets Skp2 for destruction in transforming growth factor beta-induced growth inhibition. Mol Cell Biol 2007; 27:2967-79; PMID:17283060; http://dx.doi.org/10.1128/MCB.01830-06
- Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div 2009; 4:2; PMID:19152694; http://dx.doi.org/10.1186/1747-1028-4-2
- Pesin JA, Orr-Weaver TL. Regulation of APC/C activators in mitosis and meiosis. Annu Rev Cell Dev Biol 2008; 24:475-99; PMID:18598214; http://dx.doi.org/10.1146/annurev.cellbio.041408.115949
- Pfleger CM, Lee E, Kirschner MW. Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev 2001; 15:2396-407; PMID:11562349; http://dx.doi.org/10.1101/gad.918201
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004; 428:190-3; PMID:15014502; http://dx.doi.org/10.1038/nature02330
- Bashir T, Pagano M. Don't skip the G1 phase: how APC/CCdh1 keeps SCFSKP2 in check. Cell Cycle 2004; 3:850-2; PMID:15190201; http://dx.doi.org/10.4161/cc.3.7.977
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004; 428:194-8; PMID:15014503; http://dx.doi.org/10.1038/nature02381
- Zhang L, Fujita T, Wu G, Xiao X, Wan Y. Phosphorylation of the anaphase-promoting complex/Cdc27 is involved in TGF-beta signaling. J Biol Chem 2011; 286:10041-50; PMID:21209074; http://dx.doi.org/10.1074/jbc.M110.205518
- Wasch R, Robbins JA, Cross FR. The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene 29:1-10; PMID:19826416; http://dx.doi.org/10.1038/onc.2009.325
- Chang L, Barford D. Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis. Current opinion in structural biology 2014; 29:1-9; PMID:25174288; http://dx.doi.org/10.1016/j.sbi.2014.08.003
- Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 2015; 522:450-4; PMID:26083744; http://dx.doi.org/10.1038/nature14471
- Cardozo T, Abagyan R. Druggability of SCF ubiquitin ligase-protein interfaces. Methods Enzymol 2005; 399:634-53; PMID:16338386; http://dx.doi.org/10.1016/S0076-6879(05)99042-3
- Cardozo T, Pagano M. Wrenches in the works: drug discovery targeting the SCF ubiquitin ligase and APC/C complexes. BMC Biochem 2007; 8 Suppl 1:S9; PMID:18047746
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell 2005; 20:9-19; PMID:16209941; http://dx.doi.org/10.1016/j.molcel.2005.09.003
- Sitry D, Seeliger MA, Ko TK, Ganoth D, Breward SE, Itzhaki LS, Pagano M, Hershko A. Three different binding sites of Cks1 are required for p27-ubiquitin ligation. J Biol Chem 2002; 277:42233-40; PMID:12140288; http://dx.doi.org/10.1074/jbc.M205254200
- Wang W, Ungermannova D, Chen L, Liu X. Molecular and biochemical characterization of the Skp2-Cks1 binding interface. J Biol Chem 2004; 279:51362-9; PMID:15452136; http://dx.doi.org/10.1074/jbc.M405944200
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 2001; 3:321-4; PMID:11231585; http://dx.doi.org/10.1038/35060126
- Song L, Rape M. Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol Cell 2010; 38:369-82; PMID:20471943; http://dx.doi.org/10.1016/j.molcel.2010.02.038
- Kim S, Yu H. Mutual regulation between the spindle checkpoint and APC/C. Semin Cell Dev Biol 2011; 22:551-8; PMID:21439394; http://dx.doi.org/10.1016/j.semcdb.2011.03.008
- Khattar V, Thottassery JV. Cks1: Structure, Emerging Roles and Implications in Multiple Cancers. Journal of cancer therapy 2013; 4:1341-54; PMID:24563807; http://dx.doi.org/10.4236/jct.2013.48159
- Krishnan A, Nair SA, Pillai MR. Loss of cks1 homeostasis deregulates cell division cycle. Journal of cellular and molecular medicine 2010; 14:154-64; PMID:19228269; http://dx.doi.org/10.1111/j.1582-4934.2009.00698.x
- Tomiatti V, Istvanffy R, Pietschmann E, Kratzat S, Hoellein A, Quintanilla-Fend L, von Bubnoff N, Peschel C, Oostendorp RA, Keller U. Cks1 is a critical regulator of hematopoietic stem cell quiescence and cycling, operating upstream of Cdk inhibitors. Oncogene 2015; 34:4347-57; PMID:25417705; http://dx.doi.org/10.1038/onc.2014.364
- Westbrook L, Manuvakhova M, Kern FG, Estes NR, 2nd, Ramanathan HN, Thottassery JV. Cks1 regulates cdk1 expression: a novel role during mitotic entry in breast cancer cells. Cancer Res 2007; 67:11393-401; PMID:18056467; http://dx.doi.org/10.1158/0008-5472.CAN-06-4173
- Di Fiore B, Pines J. How cyclin A destruction escapes the spindle assembly checkpoint. J Cell Biol 2010; 190:501-9; PMID:20733051; http://dx.doi.org/10.1083/jcb.201001083
- Wolthuis R, Clay-Farrace L, van Zon W, Yekezare M, Koop L, Ogink J, Medema R, Pines J. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell 2008; 30:290-302; PMID:18471975; http://dx.doi.org/10.1016/j.molcel.2008.02.027
- Morris MC, Kaiser P, Rudyak S, Baskerville C, Watson MH, Reed SI. Cks1-dependent proteasome recruitment and activation of CDC20 transcription in budding yeast. Nature 2003; 423:1009-13; PMID:12827207; http://dx.doi.org/10.1038/nature01720
- Arvai AS, Bourne Y, Hickey MJ, Tainer JA. Crystal structure of the human cell cycle protein CksHs1: single domain fold with similarity to kinase N-lobe domain. Journal of molecular biology 1995; 249:835-42; PMID:7791211; http://dx.doi.org/10.1006/jmbi.1995.0341
- Bourne Y, Watson MH, Hickey MJ, Holmes W, Rocque W, Reed SI, Tainer JA. Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle-regulatory protein CksHs1. Cell 1996; 84:863-74; PMID:8601310; http://dx.doi.org/10.1016/S0092-8674(00)81065-X
- Lecanda J, Ganapathy V, D'Aquino-Ardalan C, Evans B, Cadacio C, Ayala A, Gold LI. TGFbeta prevents proteasomal degradation of the cyclin-dependent kinase inhibitor p27kip1 for cell cycle arrest. Cell Cycle 2009; 8:742-56; PMID:19221482; http://dx.doi.org/10.4161/cc.8.5.7871
- Muggia F, Gold LI, Curtin J. Future Directions: New Targets Totowa, NJ: Humana Press, Inc., 2009.
- Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol 2007; 2:57-85; PMID:18039093; http://dx.doi.org/10.1146/annurev.pathol.2.010506.091905
- Huang KT, Pavlides SC, Lecanda J, Blank SV, Mittal KR, Gold LI. Estrogen and progesterone regulate p27kip1 levels via the ubiquitin-proteasome system: pathogenic and therapeutic implications for endometrial cancer. PLoS One 2012; 7:e46072; PMID:23029392; http://dx.doi.org/10.1371/journal.pone.0046072
- Park H, Seok JM, Yoon BS, Seong SJ, Kim JY, Shim JY, Park CT. Effectiveness of high-dose progestin and long-term outcomes in young women with early-stage, well-differentiated endometrioid adenocarcinoma of uterine endometrium. Arch Gynecol Obstet 2011; 285:473-478.
- Lecanda J, Shabtai D, Ayala A, Liarski V, Parekh TV, Gama P, Rahman MM, Mittal K, Gold LI. TGF-b prevents proteasomal degradation of p27(Kip1) by downregulation of Cks1 and Skp2 for cellular growth inhibition. Molecular Biology of the Cell 2004; 15:128
- Miyamoto T, Horiuchi A, Kashima H, Suzuki A, Yamada T, Kurai M, Konishi I, Shiozawa T. Inverse correlation between Skp2 and p27(Kip1) in normal endometrium and endometrial carcinoma. Gynecol Endocrinol 2010; 26:220-9; PMID:19724954; http://dx.doi.org/10.3109/09513590903215482
- Gold LI, Parekh TV. Loss of growth regulation by transforming growth factor-beta (TGF-beta) in human cancers: studies on endometrial carcinoma. Semin Reprod Endocrinol 1999; 17:73-92; PMID:10406078; http://dx.doi.org/10.1055/s-2007-1016214
- GLaL J.. Mechanisms ofr Cell Cycle Regulation by TGF-beta Disabled in Cancer Totowa, NJ: Humana Press, Inc, 2008.
- Akhurst RJ, Derynck R, Balmain A. TGF-beta signaling in cancer–a double-edged sword. Trends Cell Biol 2001; 11:S44-51; PMID:11684442
- Hu D, Liu W, Wu G, Wan Y. Nuclear translocation of Skp2 facilitates its destruction in response to TGFbeta signaling. Cell Cycle 2011; 10:285-92; PMID:21212736; http://dx.doi.org/10.4161/cc.10.2.14517
- Fujita T, Liu W, Doihara H, Wan Y. An in vivo study of Cdh1/APC in breast cancer formation. Int J Cancer 2009; 125:826-36; PMID:19350629; http://dx.doi.org/10.1002/ijc.24399
- Fujita T, Liu W, Doihara H, Wan Y. Regulation of Skp2-p27 axis by the Cdh1/anaphase-promoting complex pathway in colorectal tumorigenesis. Am J Pathol 2008; 173:217-28; PMID:18535175; http://dx.doi.org/10.2353/ajpath.2008.070957
- Pavlides SC, Huang KT, Reid DA, Wu L, Blank SV, Mittal K, Guo L, Rothenberg E, Rueda B, Cardozo T, et al. Inhibitors of SCF-Skp2/Cks1 E3 Ligase Block Estrogen-Induced Growth Stimulation and Degradation of Nuclear p27kip1: Therapeutic Potential for Endometrial Cancer. Endocrinology 2013; 154:4030-45; PMID:24035998; http://dx.doi.org/10.1210/en.2013-1757
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253-67; PMID:18354415; http://dx.doi.org/10.1038/nrc2347
- Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res 2011; 17:12-8; PMID:20966355; http://dx.doi.org/10.1158/1078-0432.CCR-10-0752
- Zhou Y, Ching YP, Chun AC, Jin DY. Nuclear localization of the cell cycle regulator CDH1 and its regulation by phosphorylation. J Biol Chem 2003; 278:12530-6; PMID:12560341; http://dx.doi.org/10.1074/jbc.M212853200
- Watanabe J, Watanabe K, Jobo T, Kamata Y, Kawaguchi M, Imai M, Okayasu I, Kuramoto H. Significance of p27 as a predicting marker for medroxyprogesterone acetate therapy against endometrial endometrioid adenocarcinoma. Int J Gynecol Cancer 2006; 16 Suppl 1:452-7; PMID:16515645; http://dx.doi.org/10.1111/j.1525-1438.2006.00418.x
- Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell 2006; 124:89-103; PMID:16413484; http://dx.doi.org/10.1016/j.cell.2005.10.032
- Hall MC, Warren EN, Borchers CH. Multi-kinase phosphorylation of the APC/C activator Cdh1 revealed by mass spectrometry. Cell Cycle 2004; 3:1278-84; PMID:15467459; http://dx.doi.org/10.4161/cc.3.10.1153
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M. Mammalian Cdh1/Fzr mediates its own degradation. Embo J 2004; 23:1619-26; PMID:15029244; http://dx.doi.org/10.1038/sj.emboj.7600149
- Pines J. Cell cycle: reaching for a role for the Cks proteins. Curr Biol 1996; 6:1399-402; PMID:8939596; http://dx.doi.org/10.1016/S0960-9822(96)00741-5
- Keller UB, Old JB, Dorsey FC, Nilsson JA, Nilsson L, MacLean KH, Chung L, Yang C, Spruck C, Boyd K, et al. Myc targets Cks1 to provoke the suppression of p27Kip1, proliferation and lymphomagenesis. Embo J 2007; 26:2562-74; PMID:17464290; http://dx.doi.org/10.1038/sj.emboj.7601691
- O'Hagan RC, Ohh M, David G, de Alboran IM, Alt FW, Kaelin WG, Jr., DePinho RA. Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev 2000; 14:2185-91; PMID:10970882; http://dx.doi.org/10.1101/gad.827200
- Lan Y, Zhang Y, Wang J, Lin C, Ittmann MM, Wang F. Aberrant expression of Cks1 and Cks2 contributes to prostate tumorigenesis by promoting proliferation and inhibiting programmed cell death. Int J Cancer 2008; 123:543-51; PMID:18498131; http://dx.doi.org/10.1002/ijc.23548