
ABSTRACT
The clonal expansion of activated T cells is pivotal for the induction of protective immunity. Protein phosphatase 4 (PP4) is a ubiquitously expressed serine/threonine phosphatase with reported functions in thymocyte development and DNA damage responses. However, the role of PP4 in T cell immunity has not been thoroughly investigated. In this report, our data showed that T cell-specific ablation of PP4 resulted in defective adaptive immunity, impaired T cell homeostatic expansion, and inefficient T cell proliferation. This hypo-proliferation was associated with a partial G1-S cell cycle arrest, enhanced transcriptions of CDK inhibitors and elevated activation of AMPK. In addition, resveratrol, a known AMPK activator, induced similar G1-S arrests, while lentivirally-transduced WT or constitutively-active AMPKα1 retarded the proliferation of WT T cells. Further investigations showed that PP4 co-immunoprecipitated with AMPKα1, and the over-expression of PP4 inhibited AMPK phosphorylation, thereby implicating PP4 for the negative regulation of AMPK. In summary, our results indicate that PP4 is an essential modulator for T cell proliferation and immune responses; they further suggest a potential link between PP4 functions, AMPK activation and G1-S arrest in activated T cells.
Introduction
Upon the recognition of antigens, naïve T cells are signaled to initiate a series of genetic and metabolic re-programming that induces their expansion and differentiation into effector T cells to provide protective immunity for the host. Once pathogens are cleared and the immune responses subside, this pool of effector T cells rapidly shrinks via programmed cell death. This pathogen-specific expansion and contraction of immune cells is the hallmark of the immune system.Citation1,2 Initial signals from the antigen-presenting cells trigger cascades of pathways to induce the transcriptional activation of cytokines, growth factors and cell cycle regulators in activated T cells. Feedback amplification by these factors then sustains the expression of pro-expansion genes and permits the continuous proliferation of activated T cells.Citation1 The signals from IL-2 serve as such a positive feedback loop by inducing the expression of high affinity IL-2 receptor subunit CD25/IL-2Rα, as well as IL-2 itself, to facilitate T cell proliferation and differentiation.Citation3
In parallel to these genetic activations, recent findings also suggest that T cells must adapt to metabolic changes so that they can transit rapidly from the resting state to become vigorously-proliferating cells.Citation4,5 These changes include enhanced nutrient uptake and biosynthesis, the switch from the tricarboxylic acid cycle to aerobic glycolysis for ATP generation, and the induction of limited autophagy. These metabolic changes, which often do not require transcriptional regulations, then help set the stage for activated naïve T cells to expand and differentiate into effector T cells.Citation4,5 Understanding how these metabolic changes are regulated in activated T cells is thus pivotal for studying T cell immune responses.Citation4,5
AMP-activated protein kinase (AMPK) and mechanistic target of rapamycin (mTOR) are 2 important serine/threonine kinases that are responsible for regulating cellular metabolism. mTOR is activated by growth factors, and can modulate biosynthesis processes such as protein translation, lipogenesis and mitochondria metabolism to promote cellular expansion.Citation6,7 In contrast, AMPK, as an energy sensor, is activated by AMP and suppressed by ATP; additional signals, such as Ca2+ flux and reactive oxygen species (ROS) can also activate AMPK.Citation8,9 Activated AMPK then acts by inhibiting mTOR functions or cell cycle progression to negatively regulate proliferation.Citation10 Furthermore, AMPK can also phosphorylate Atg1/ULK1 to promote autophagy as a means of recycling biosynthesis materials under nutrient-depleted conditions.Citation11,12 In this regard, resveratrol (Resv), a natural phenol, has been shown to activate AMPK to induce autophagy Citation13 and inhibit T cell proliferation.Citation14,15
Protein phosphatase 4 (PP4) is a ubiquitously expressed serine/threonine phosphatase encoded by the ppp4c gene. The genomic deletion of the ppp4c gene results in embryonic lethality, hinting that PP4 may be essential for cell expansion and differentiation.Citation16 T cell-specific ablation of PP4 by the proximal Lck promoter-driven Cre recombinase transgene (Lck-cre) causes severe thymocyte development blocks and induces peripheral lymphopenia.Citation16 In contrast, knockout of PP4 by the CD4 promoter-driven Cre recombinase transgene (CD4-cre) does not significantly impact thymocyte differentiation, but partially impairs regulatory T cell functions to induce the onset of spontaneous colitis.Citation17 Recently, PP4 has also been implicated in DNA damage response via its ability to either permit cell cycle reentry,Citation18 dephosphorylate γH2AX,Citation19,20 regulate the activity of KAP1,Citation21 or control cell cycles in Drosophila Citation22 or yeast Citation23; however, its role in regulating mammalian cell proliferation has not been thoroughly investigated. Finally, it is worth noting that okadaic acid (OA), which is generally recognized as a specific inhibitor of PP2A, actually also suppresses PP4 activity with equal Citation24 or better Citation25 efficacy; these results then raise the possibility that many biological processes, such as IL-2 signaling modulation,Citation26,27 AMPK activation Citation28 and the regulation of T cell proliferation,Citation29 that have been linked to PP2A via OA treatments may actually be attributed to the functions of PP4.
Our previous characterizations of mice with CD4-cre mediated deletion of the ppp4c gene (CD4cre:PP4f/f) revealed a reduction in the number of peripheral CD4 and CD8 T cells.Citation17 In this report, we further showed that the T lymphopenia in CD4cre:PP4f/f mice could be attributed to the reduced homeostatic capacity and hypo-proliferation of PP4-deficient T cells. This T cell hypo-proliferation was not caused by defective IL-2 production or signalings. Instead, PP4 deficiency resulted in a partial G1-S cell cycle block that was associated with AMPK hyper-activation.
Results
Defective T cell immunity and T-dependent humoral responses in CD4cre:PP4f/f mice
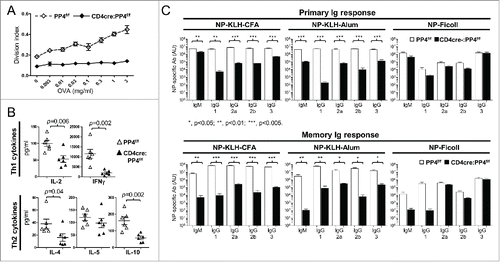
PP4 was reported to be essential for pre-TCR signaling Citation16 and T cell survival.Citation30 In addition, our previous report showed that CD4cre:PP4f/f mice suffered from αβ T cell lymphopenia and exhibited reduced KLH T cell responses.Citation17 To further investigate the functions of PP4 in peripheral T cells, we immunized 6–8 wk old WT or CD4cre:PP4f/f mice with OVA/CFA and harvested the draining LN T cells for OVA re-stimulation in vitro. The results showed that T cells from CD4cre:PP4f/f mice were completely unresponsive (), similar to our previous report analyzing KLH/CFA immunization.Citation17 Analyses of cytokine secretion in the re-stimulated cultures revealed significantly-reduced secretion of IL-2, IFN-γ, IL-4 and IL-10 by PP4-deficient T cells (p = 0.002–0.04, ). When primary and memory humoral responses were compared between PP4f/f and CD4cre:PP4f/f littermates following NP-KLH/CFA, NP-KLH/alum or NP–ficoll immunization, serum ELISA results from the NP-KLH/CFA or NP-KLH/alum immunizations showed that T-dependent antibody responses were severely impaired by PP4 deficiency (p < 0.001–0.05 for all Ig isotypes, , top row); similar results were also observed in the memory responses (p < 0.001–0.05 for all Ig isotypes, , bottom row). In contrast, T-independent antibody responses induced by NP-ficoll were either unaltered, or only marginally affected (IgM and IgG1 memory response, p > 0.05, , bottom row) by PP4 ablation. The severely hampered T-dependent immune responses in the CD4cre:PP4f/f mice thus suggest that PP4 is essential for the optimal induction of T cell immunity.
Figure 1. CD4cre:PP4f/f mice exhibit defective T-dependent immune responses in vivo. (A) Mice at 6–8 wk age were immunized with OVA/CFA emulsion in the foot pad; one wk later lymphocytes from the draining LN were assessed in vitro by CFSE dye dilution for OVA-induced T cell proliferation (n = 8). (B) Day 3 culture supernatants from cells re-stimulated with 3 μg/ml OVA in (A) were subjected to multiplex assay to measure Th1/Th2 cytokines secretion (n = 6). (C) Mice at 6–8 wk age were immunized i.p. with the indicated epitope/antigen/adjuvant, and their sera were collected on d 21 for primary Ig responses (top panels). Mice were immediately boosted by immunization and their sera collected again on d 35 for memory Ig response (bottom panels). (n = 3–4). AU, arbitrary unit. *, p < 0.05; **, p < 0.01; ***, p < 0.005. See Supplemental Figure S1A for flow cytometry gating strategies.
PP4 ablation impedes T cell homeostatic expansion in vivo
Our previous report showed that CD4cre:PP4f/f mice exhibited partial αβ T lymphopenia.Citation17 Together with the defective T cell responses (), these observations raise the possibility that potential alterations in T cell expansion may contribute to these phenotypes. Support for this possibility was found when we assessed the efficiency of ppp4c gene deletion in peripheral T cells. By using qPCR to quantitate the floxed region and flanking control region of the ppp4c gene ( and Citation17), we found that the floxed ppp4c exon was deleted in ∼90% of splenic CD4 T cells and ∼75% of splenic CD8 T cells from 6 wk and 12 wk old CD4cre:PP4f/f mice (). However, in 24 wk old CD4cre:PP4f/f mice only ∼80% of the ppp4c gene was deleted in CD4 T cells, and the deletion efficiency dropped to ∼25% in CD8 T cells (p = 0.02, ); similar results were also observed in cells from the mesenteric lymph node (MLN) (p = 0.01, ). The control CD4−CD8− populations showed no change in PP4 deletion efficiency (, right panel). This age-dependent reduction of ppp4c deletion efficiency in the CD4 and CD8 T populations is consistent with the possibility that PP4-deficient T cells, when competing with PP4-sufficient T cells that have escaped from the CD4-cre-mediated deletion in the thymus, exhibit a proliferation disadvantage in the periphery.
Figure 2. The ablation of PP4 impairs T cell expansion. (A) Schematics of the genomic qPCR assay for quantifying ppp4c deletion efficiency. a-d, PCR primers. Triangle, lox-P sites. (B) CD4, CD8 or control CD4−CD8− splenocytes or MLN cells were sorted by flow cytometry from mice at different ages; DNA extracted from these cells was analyzed by qPCR as in (A) for assessing the deletion efficiency of ppp4c (n = 2–3). (C) Schematics of the homeostatic expansion assay (left panel) and the representative CD90.2 histogram of CD4 splenocytes from the recipient (right panel) are shown. (D-E) WT (CD90.2−) or CD4cre:PP4f/f (CD90.2+) total T cells were purified from pooled spleen and LN by MACS, mixed at a 1:1 ratio (day 0), labeled with CFSE, and transferred into RAG1−/− mice. At day 1 or day 8 post-transfer, spleen, ALN or MLN cells from the recipients were analyzed by flow cytometry for the percentages of WT or CD4cre:PP4f/f donor cells and their respective CFSE dye-dilution patterns. The relative percentages of WT and CD4cre:PP4f/f donor cells (left panels) in gated CD4+ (D) or CD8+ (E) populations are shown. Representative CFSE dye dilution profiles of WT and CD4cre:PP4f/f donor cells are also shown (right panels) (n = 3). See Supplemental Figure S1A–B for flow cytometry gating and sorting strategies.
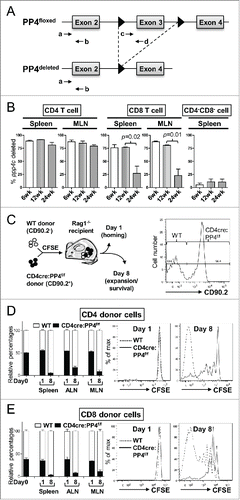
To investigate whether PP4-deficient T cells indeed expand less efficiently than WT T cells, we mixed WT (CD90.2−) and CD4cre:PP4f/f (CD90.2+) T cells, loaded them with CFSE, and adoptively transferred them into RAG1−/− recipients (). Flow cytometry results on day 1 post-transfer showed that the ratios of WT and PP4-deficient CD4 (, left panels) or CD8 (, left panels) T cells were similar in the spleen, auxiliary LN (ALN) and MLN, suggesting that the lack of PP4 does not impact the short-term survival or homing ability of peripheral T cells. More importantly, by day 8 the percentages of WT CD4 and CD8 T cells in the recipient mice increased significantly to ∼90% (, left panels). CFSE dye-dilution patterns revealed that, while WT CD4 and CD8 T cells proliferated vigorously, PP4-deficient T cells barely expanded (, right panels). These results thus suggest that PP4 is an essential regulator for the homeostatic expansion of peripheral T cells. In this regard, they also imply that the partial lymphopenia Citation17 and T immune-deficiency () in the CD4cre:PP4f/f mice may be attributed to reduced T cell proliferation.
PP4 deficiency induces G1-S blocks and retards T cell proliferation via cell-intrinsic pathways
To test whether PP4 deficiency indeed impacts T cell proliferation, T cells from 6–8 wk old PP4f/f and CD4cre:PP4f/f littermates were purified and activated in vitro by anti-CD3ε and anti-CD28 antibodies. The characterization of proximal signaling events by western blotting and Ca2+ flux analyses revealed no significant defect in the activation of ZAP70, Akt, ERK, JNK, p38 and IKKβ (Fig. S2A) or the release of Ca2+ into the cytoplasm (Fig. S2B). This is different from a previous report showing that pre-TCR signaling is altered in PP4-deficiency thymocytes,Citation16 but may simply represent differential requirements of PP4 for pre-TCR and TCR signalings. CFSE dye-dilution results for the activated T cells showed that PP4-deficient CD4 T cells were significantly hypo-proliferative as early as day 2 (p = 0.003–0.008, ). Similar results were observed for CD8 T cells; however, CD8-related data were not shown hereon due to potential artifacts introduced by the reduced PP4 deletion efficiency in the CD8 T population ().
Figure 3. The hypo-proliferation of PP4-deficient CD4 T cells coincides with a partial G1-S block, and is mediated primarily via cell-intrinsic mechanisms. (A-B) Total T cells were purified by MACS, labeled with CFSE, stimulated with a non-saturating dose of 1.6 μg/ml anti-CD3ε and anti-CD28, and analyzed daily by flow cytometry. Representative CFSE profiles (panel A) and division indexes calculated by the FlowJo software (panel B) of gated CD4 cells are shown (n = 6–7). (C-D) MACS-purified T cells were activated as in panel A in the presence of 10 μM BrdU; at the specified time, cells were fixed and analyzed for cell cycle status by intracellular staining of BrdU and 7AAD. Representative FACS plots and gate nomenclatures (panel C), as well as statistical analyses of cell cycle status (panel D) from CD4 T cells are shown (n = 4–5). (E-F) Schematics and results of the competitive proliferation assay. MACS-purified WT and CD4cre:PP4f/f T cells were mixed at 1:1 ratio, labeled with CFSE, and activated and analyzed as in panel A (panel E). The percentages of WT or CD4cre:PP4f/f T cells in gated CD4 populations are shown (n = 6–7) (panel F). See Supplemental Figure S1A, -C and-D for flow cytometry gating strategies.
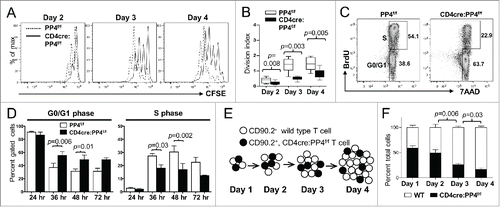
To gain more insight into the hypo-proliferation of PP4-deficient T cells, we examined their apoptosis induction and cell cycle progression. AnnexinV/7AAD staining results of anti-CD3ε and anti-CD28-activated T cells showed that PP4 deficiency did not significantly change the induction of apoptosis (p > 0.05, Fig. S3A); in addition, the transcription of apoptosis-related genes such as Bim, Bax and Puma was also unaltered by the ablation of PP4 (Fig. S3B), hinting that the reduced expansion of PP4-deficient T cells was not caused by enhanced apoptosis. In contrast, BrdU labeling of activated T cells () showed that PP4-deficient CD4 T cells exhibited a partial block at the G1-S transition that became appreciable at 36 hr following TCR stimulation: this was demonstrated by the increased percentages of G0/G1 cells (p = 0.006–0.01, , left panel) and the decreased percentages of cells in the S phase (p = 0.002–0.03, , right panel) in the PP4-deficient populations.
Lastly, to test whether the defective T cell proliferation was an cell-intrinsic property of PP4-deficient T cells, we performed a competitive proliferation assay by mixing equal numbers of WT and PP4-deficient T cells and stimulating them together in vitro (). The results showed that the percentages of PP4-deficient CD4 T cells decreased gradually from ∼55% to ∼20% (p = 0.006–0.03, ). Combined with the mixed adoptive transfer experiments (), these results indicate that PP4 deficiency induces T cells hypo-proliferation predominantly through cell-intrinsic regulations. In addition, the unaltered proximal TCR signaling in PP4-deficient T cells further indicates that these regulations are likely enforced via distal, downstream pathways.
Reduced IL-2 production or signalings is not the cause for the T cell hypo-proliferation
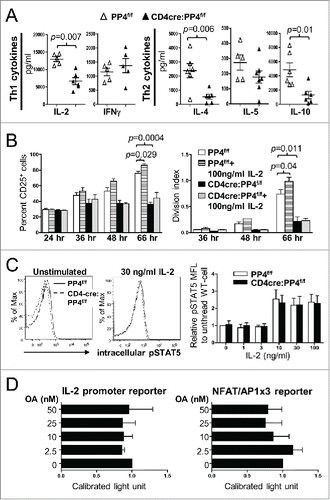
IL-2 is an important cytokine that facilitates the progression from the G1 phase into the S phase of the cell cycle.Citation3 Treatment with OA (and the resulting suppression of PP2A, PP4 and PP6) has been reported to down-regulate the mRNA level of IL-2 Citation25 through multiple pathways.Citation27,31 In addition, our data showed that activated PP4-deficient T cells produced significantly less IL-2 (p = 0.007) (as well as IL-4 and IL-10) (), and exhibited reduced induction of the high-affinity IL-2 receptor subunit, IL-2Rα/CD25 (66 hr, p = 0.0004, ). To test whether PP4 deficiency inhibits IL-2 secretion or signalings to retard the proliferation of PP4-deficient T cells, we treated activated T cells with exogenous IL-2. The results showed that exogenous IL-2 enhanced the induction of CD25 in WT CD4 T cells (66 hr, p = 0.029, left panel, ) and promoted their proliferation (p = 0.04, right panel, ). In contrast, PP4-deficient CD4 T cells were irresponsive to the IL-2 treatment (p > 0.05, ), suggesting that the hypo-proliferation of PP4-deficient T cells is not due to reduced initial production of IL-2. When we stimulated CD25+ WT or CD4cre:PP4f/f T cells with titrating doses of IL-2, we found that IL-2 induced similar extents and kinetics of STAT5 phosphorylation in both populations (), suggesting that proximal IL-2 signaling was also unaffected by PP4 deficiency. Lastly, when 2 luciferase reporter assays were employed to test whether PP4 regulates IL-2 transcriptions, we found that the inhibition of PP2A/PP4/PP6 by OA failed to reduce the activity of IL-2 promoter () even at a relatively high dose of 50 nM,Citation25 hinting that PP4 deficient may not significantly alter IL-2 transcription. Combined with the mixed proliferation data (), these results suggest that PP4 deficiency likely did not impair the signalings or production of IL-2 to retard T cell proliferation. On the contrary, the hypo-proliferation of PP4-deficient T cells may have contributed to the reduced secretion of IL-2.
Figure 4. Reduced IL-2 production or signalings is not the primary cause for the hypo-proliferation of PP4-deficient T cells. (A) Day 3 culture supernatants from were analyzed by multiplex assays for the secretion of various Th1/Th2 cytokines (n = 5–6). (B) MACS-purified, CFSE-loaded T cells were activated by sub-optimal 0.5 μg/ml plate-bound anti-CD3ε and anti-CD28 in the presence or absence of exogenous IL-2; this was then followed by the analyses of CD25 induction (left panel) or proliferation (right panel) on gated CD4 populations (n = 2–3). (C) MACS-purified T cells were activated by 1.6 μg/ml anti-CD3ε and anti-CD28 for 2 d, washed, and rested for 4 hr. Rested WT and CD4cre:PP4f/f cells were then treated with titrating doses of IL-2 and stained intracellularly for phospho-STAT5-Y694. Representative flow cytometry plots (left panels) and statistical analyses of the mean fluorescence levels (right panel) are shown (n = 2–4). (D) JTAg cells were transfected with the respective luciferase reporter plasmids and control tdTomato plasmid by electroporation; cells were stimulated 24 hr later with PMA/ionomycin and various doses of OA for 6 hr prior to luciferase activity measurement, as well as FACS analyses for assessing the transfection efficiency by tdTomato fluorescence. Luciferase activity after compensating for transfection efficiency and normalized to the no-OA samples (Calibrated light unit) are shown (n = 3). See Supplemental Figure S1A and -E for flow cytometry gating strategies.
The partial G1-S arrest coincides with increased expressions of CDK inhibitors Ink4a and Ink4b
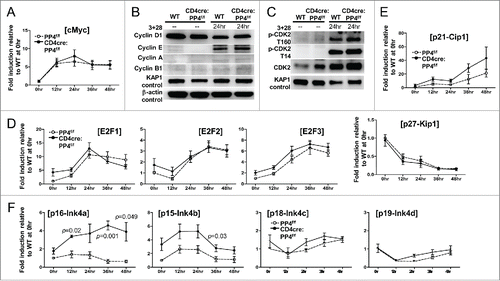
To see if PP4 ablation caused the partial G1-S block by altering the expression or activation of various cell cycle regulators, we performed qPCR and Western blotting analyses on activated WT and PP4-deficient T cells. The qPCR results revealed similar kinetics of cMyc expression between the 2 populations (). Western blotting analyses further showed that the inductions of cyclin-D1, -E, -A and –B1 (), as well as the activation of CDK2 (assessed by both the activating Thr-160 and the inhibitory Thr-14 phosphorylation,Citation32 ), were all unaltered in PP4-deficient T cells at 24 hr post-stimulation. Consistent with the western analyses results, the induction of CDK2 target genes, such as E2F1, E2F2 and E2F3, remained largely unaffected by PP4 deficiency (). The transcription of the CDK inhibitor p27-Kip1 was also similar between WT and PP4-deficient T cells, but the expression of its family member, p21-Cip1, was slightly enhanced (p > 0.05) in PP4-deficient T cells (). More importantly, qPCR results revealed significantly-increased expressions of p16-Ink4a (p = 0.001–0.049) and p15-Ink4b (p = 0.03) () in PP4-deficient T cells, while normal levels of p18-Ink4c or p19-Ink4d were observed (). The enhanced expression of p16/Ink4a and p15/Ink4b may thus contribute to the hypo-proliferation in PP4-deficient T cells; however, at this point we do not know their targets or their function mechanisms.
Figure 5. The partial G1-S block induced by PP4 deficiency is associated with enhanced expression of CDK inhibitors p16/Ink4a and p15/Ink4b. (A, D-F) RNA was collected from T cells activated as in at the specified time for qPCR analyses. Fold inductions relative to unstimulated WT T cells, after compensating for signals from β–actin, are shown for the respective genes (n = 3–7). (B-C) MACS-purified T cells were activated as above with their lysates collected at the indicated time for western blot analyses. Representative protein gel blotting results are shown (n = 3–5). 3 + 28, anti-CD3ε+anti-CD28 stimulation.
AMPK hyper-activation induced by PP4 deficiency may contribute to T cell hypo-proliferation
Recent reports suggest that naïve T cells must pursue metabolic adaptations in order to sustain the vigorous proliferation induced by antigen activation.Citation4,5 During this metabolic adaptation, AMPK (as AMPKα1, the predominantly-expressed isoform in T cells Citation33) has been shown to play an important role by regulating the activity of mTOR and the homeostasis of ROS.Citation34,35 In this regard, we found that AMPK activation (as marked by its Thr-172 phosphorylation) was stronger in both naïve and activated PP4-deficient T cells (). The activation of mTOR, however, was mostly unaffected by the ablation of PP4 (), as was the phosphorylation of mTOR substrates S6K and 4E-BP1 (). When T cells were treated with resveratrol (Resv), a known AMPK activator Citation13 and inhibitor of T cell proliferation,Citation14,15 we found that Resv was able to induce a partial G1-S arrest in WT CD4 T cells similar to that of untreated PP4-deficient T cells (p < 0.01–0.05, ); yet it failed to further exacerbate the existing G1-S arrest in PP4-deficient T cells (p > 0.05, ). This lack of additive or synergizing effects between Resv and PP4 deficiency suggest that they may induce the G1-S blocks via a common pathway. In parallel, when constitutively-active (CA)-AMPKα1 (T183D point mutation and Y323 c-terminal truncation,Citation36 ) was lentivirally transduced (with a bicistronic CD90.1 infection marker, ) into WT T cells, we found that CA-AMPKα1 inhibited the proliferation of infected CD90.1+ CD4 T cells (p = 0.044, , left panel); interestingly, the overexpression of WT AMPKα1 also inhibited the proliferation of infected CD4 T cell (p = 0.001, right panel), suggesting that AMPK over-expression may be sufficient to interfere with T cell proliferation. More importantly, similar to the results from Resv treatments (), PP4-deficient T cells again failed to respond to either CA- or WT-AMPKα1 transfection (), thereby supporting an overlapping functional pathway between AMPK activation and PP4 deficiency for the regulation of T cell proliferation.
Figure 6. AMPK hyper-activation is induced by PP4 ablation and correlates with retarded T cell proliferation. (A-B) MACS-purified T cells were activated and analyzed by western blot analyses as in for AMPK activation (panel A), or mTOR activation and mTOR substrate phosphorylation (panel B). Representative protein gel blotting results are shown (n = 3). 3+28, anti-CD3ε+anti-CD28 stimulation. (C) MACS-purified T cells were activated as in in the presence or absence of 20 μM Resv and 10 μM BrdU. After 48 hr, cells were fixed and analyzed for cell cycle status by intracellular staining of BrdU and 7AAD. Statistical analyses results of gated CD4 T cells are shown (n = 4–5). (D) Schematics of WT- and CA-AMPKα1. (E) Schematics of the bicistronic lentiviral expression vector. IRES, internal ribosome entry site. Pbsd-CD90.1, blasticidin-resistant and CD90.1 fusion gene. (F) MACS-purified T cells were CFSE-loaded and activated as in in the presence of control lentivirus (vector, with CD90.1 infection marker only) or lentivirus with CA- (left panel) or WT-AMPKα1 (right panel) plus CD90.1 marker for 48 hr. Division indexes of the infected, CD90.1+ CD4 T cells are shown (n = 3–5). (G) Myc-tagged PP4 and flag-tagged AMPKα1 or control flag vector were transiently co-expressed in HEK-293T cells for 24 hr; this was then followed by lysate collection and anti-flag immunoprecipitation. Representative western blotting results from pre-immunoprecipitated lysates (IB, top half) and anti-flag-precipitated fractions (IP-Flag, bottom half) are shown (n = 3). (H) HEK-293 clones with tetracycline-inducible expression of myc-tagged PP4 (293-TO-PP4-Myc, left panel) or GFP (293-TO-GFP, right panel) were treated with 2 μg/ml tetracycline (Tet-on) for 24 hr; this was then followed by the addition of 1 mM H2O2 for the indicated time. Representative protein gel blot results from 3 independent clones are shown (n = 3). See Supplemental Figure S1F for flow cytometry gating strategies.
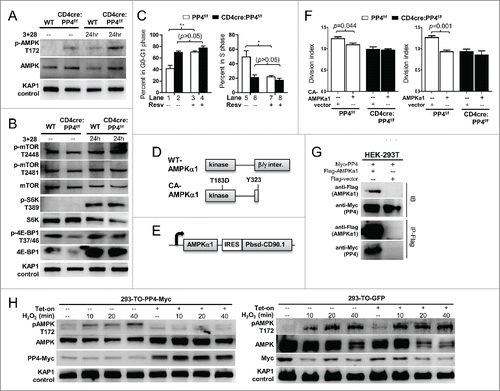
As a serine/threonine phosphatase, PP4 may dephosphorylate AMPK to modulate its activity. To test this possibility, we transiently co-expressed PP4 and AMPKα1 in HEK-293T cells, and found that PP4 could be efficiently co-immunoprecipitated with AMPKα1 (). We next generated HEK-293 clones with tetracycline-inducible expression of PP4 (293-TO-PP4-Myc) or control GFP (293-TO-GFP), and used exogenous hydrogen peroxide to induce AMPK phosphorylation. While hydrogen peroxide efficiently induced Thr-172 phosphorylation of endogenous AMPK in 293-TO-PP4-Myc cells (, left panel), pre-treatment with tetracycline and the resulting induction of PP4 significantly suppressed AMPK activation (, left panel); meanwhile, tetracycline treatments and GFP induction did not alter AMPK phosphorylation in the control 293-TO-GFP cells (, right panel), thereby excluding direct suppressions of AMPK by tetracycline. These results, when combined with the enhanced AMPK activation in PP4-deficient T cells (), suggest that PP4 may serve as a negative regulator of AMPK.
Discussion
While PP4 has been proposed to participate in thymocyte maturation Citation16 and T cell survival,Citation30 functional analyses of PP4-deficient T cells are either prohibited due to the severe lymphopenia in vivo,Citation16 or are shadowed by the use of PP4 overexpression or knockdown in vitro.Citation30 In contrast, the conditional deletion of the ppp4c gene by CD4-cre in this report clearly demonstrates that PP4 deficiency results in defective T cell homeostatic expansion, cytokine production, and impaired T-dependent immunity. Using this genetic model, we have thus provided unambiguous in vitro and in vivo evidence that PP4 serves as an essential positive regulator for T cell proliferation and adaptive immunity.
How can PP4 regulate AMPK activation? Such a regulation may exert its effects at 2 levels. In the upstream, AMPK has been reported to be phosphorylated and activated by LKB1, TAK1 or CaMKKβ, with different stimuli controlling the activity of individual kinases.Citation4,5 In these regards, neither Ca2+ nor proximal TCR signaling is altered by PP4 ablation (Supplemental Fig. S2A–B); meanwhile, PP4 has not been implicated in existing literatures for the direct modification of these kinases. PP4 is thus unlikely to modulate AMPK via these upstream pathways. In contrast, PP4, as a serine/threonine phosphatase, is certainly equipped to directly dephosphorylate AMPK to inactivate its kinase activity, similar to what was reported for PP2A,Citation37 PP2C Citation38 and α–SNAP.Citation39 Our co-immunoprecipitation () and, to a certain extent, tetracycline-induced PP4 over-expression () results support this possibility. We thus favor the hypothesis that PP4 may directly modulate AMPK activity by inducing its dephosphorylation.
The next lingering question is: how does PP4 deficiency, and consequently AMPK hyper-activation, cause the partial G1-S block in activated T cells? Recent reports showed that the activation of AMPK, either by enforced expression of LKB1,Citation40 by the expression of constitutively-active AMPK,Citation36 or by the chemical activators metformin and AICAR,Citation41 can induce G1-S blocks in tumor cells by altering the expressions of p21, p27, p53 or Cyclin D1. However, our analyses only found enhanced expressions of the CDK inhibitors p16/Ink4a and p15/Ink4b in activated PP4-deficient T cells (and data not shown). We believe that this discrepancy may be explained by the differential requirements of AMPK in tumor cells and activated primary T cells.Citation34,35 While tumor cells often constitutively turn on AMPK to adapt aerobic glycolysis for optimal expansion, primary T cells only transiently turn on AMPK following TCR activation.Citation33 The hyper-activation of AMPK may thus have different effects in tumor cells and activated T cells, so that the same G1-S block can be mediated via distinct pathways. It will be interesting to study how AMPK hyper-activation can induce the expression of p16/Ink4a and p15/Ink4b (located together in the INK4b-ARF-INK4a locus, reviewed inCitation42) in activated T cells to modulate T cell proliferation.
Our characterizations of PP4-deficient T cells show that they exhibit normal TCR signalings and Ca2+ flux, can be induced to express CD25/IL2-Rα, and respond to IL-2 stimulation by activating STAT5. However, they produce significantly lower amounts of IL-2, are partially blocked at the G1-S checkpoint, and fail to mount T-dependent immune responses. These characteristics are reminiscent of T cell anergy, an unresponsive state of T cells that can arise in the absence of proper co-stimulations.Citation43 Interestingly, enhanced AMPK activation, which we observed in PP4-deficient T cells, has also been implicated in the induction of anergy.Citation43,44 The results in this report then lead us to speculate that PP4 may serve as a novel mediator for the anergy avoidance signal,Citation4,5 so that T cells receiving proper TCR and co-stimulatory signals may proliferate and differentiate normally. In this regard, 2 observations distinguish our CD4cre:PP4f/f mice from the current paradigm of anergy inductionCitation43: first, exogenous IL-2 fails to rescue PP4-deficient T cells from their unresponsive state (); second, despite the enhanced AMPK activation, mTOR activity is not significantly suppressed in PP4-deficient T cells (). PP4 may thus represent a novel IL-2/mTOR-independent point of regulation for anergy prevention in activated T cells. Further investigations are required to test this possibility.
Materials and methods
Mice
PP4f/f,Citation16 CD4-cre transgenic Citation45 and RAG1−/− Citation46 mice have been described. PP4f/f mice were crossed with the CD4-cre transgenic mice to generate CD4cre:PP4f/f mice (CD90.1−/CD90.2+), in which ppp4c were specifically deleted in T cells.Citation17 C57Bl/6 CD90.1 congenic mice (CD90.1+/CD90.2−) were obtained from the Jackson Laboratory. Mice were housed under specific pathogen-free conditions at the Laboratory Animal Center of the National Health Research Institutes (NHRI). All animal experimental procedures were preapproved by the NHRI's Institutional Animal Care and Use Committee.
PCR, quantitative-PCR (qPCR), and plasmid constructions
Genotyping PCR for the CD4-cre transgene and PP4f allele was performed as described previously, Citation17 as was the qPCR measurement of ppp4c gene deletion efficiency.Citation17 For RT-qPCR, total RNA were extracted from activated T cells and converted into cDNA with standard protocols. qPCR was performed with Luminaris Color HiGreen High ROX qPCR Master Mix (Thermo Scientific, K0361) on Realplex4 with Mastercycler ep realplex software (Eppendorf). For cloning mouse PP4 cDNA, RT-PCR was performed on mouse splenic cDNA, with the product cloned via AsiSI/MluI into pCMV6-AC-Myc (Origene) and validated by sequencing. For cloning mouse AMPKα1 cDNA, RT-PCR was performed on mouse brain cDNA, with the product cloned via AsiSI/MluI into pCMV6-AC-DDK(flag-tag) (Origene) and validated by sequencing. CA-AMPKα1 (T183D point mutation and Y323 truncation; equivalent to T172D and Y312 of AMPKα2 Citation36) was then constructed through PCR-based point mutation and truncation with standard protocols, and validated similarly. The CD90.1+ lentiviral expression backbone was generated by ligating the AscI/StuI fragment (containing the blasticidin resistant gene-CD90.1 fusion gene) of pLKO_TRC024 into AscI/StuI digested pLAS2w.Pbsd (both from National Core Facility for Manipulation of Gene Function by RNAi, miRNA, miRNA sponges, and CRISPR / Genomic Research Center) to replace the single blasticidin-resistant gene. WT- or CA-AMPKα1 was then lifted by AsiSI/SmaI from the pCMV6-AC-DDK backbones into the AsiSI/PmeI digested lentivirus backbone. Oligonucleotide sequences are listed in Supplemental Table 1, including the Ink4 family primers.Citation47 PCR thermocycler conditions are available upon request.
Flow cytometry
Antibodies used for flow cytometry, including CD4, CD8, CD25, CD90.1, CD90.2, TCRβ, phospho-STAT5 Y694, BrdU and AnnexinV conjugated with various fluorescent dyes, were purchased (BioLegend or BD Biosciences). Catalog numbers of the antibodies are available upon request. Seven-aminoactinomycin D (7AAD) and BrdU were purchased (Sigma-Aldrich, A9400 and B9285). BrdU/7AAD staining was performed using Cytofix/Cytoperm buffers (BD Biosciences, 554714) following the manufacturer's instructions. The CellTrace/CFSE cell proliferation kit was purchased (Thermo Fisher, C34554), and was loaded into cells at 10 μM at 37°C for 10 min. For intracellular phospho-STAT5 staining, naïve T cells were activated by anti-CD3ε and anti-CD28 (BD Biosciences) for 48 hr, rested for 4 hr, stimulated with serially diluted IL-2 (PeproTech, 212–12) and stained for phospho-STAT5 (Tyr-694) with Phosflow Perm Buffer III (BD Biosciences, 5585050) following the manufacturer's instructions. For Ca2+ flux, mouse splenocytes were loaded with 2.5 nM of the calcium indicator Fluo-4 (Thermo Fisher, F-14201) at 37°C for 45 min in dark, washed with complete media, rested for 30 min at 37°C, and stained with surface CD4 and CD8 as well as 10 μg/ml biotinylated anti-CD3ε and anti-CD28 (BioLegend) for 10 min on ice. Cells were then washed twice and resuspended in 100 μl of complete media. To measure Ca2+ flux, baseline signals were recorded for 1 min on flow cytometry at 37°C; this was then followed by the addition of 100 μl of 2 μg/mL crosslinking streptavidin (Sigma-Aldrich, S4762) and signal-recording for 10 min; finally 2.5 μg/mL ionomycin (Sigma-Aldrich, I9657) was added to establish the maximum Ca2+ flux. Flow cytometry results were captured on FACSCanto II (BD Biosciences), and the results were analyzed by FlowJo software (Tree Star). Division indexes were calculated by the FlowJo software based on the CFSE-dye-dilution patterns. In short, each CFSE peak/cell population was first assigned with a number of pursued cellular divisions per cell based on its relative position to the rightmost peak, which represented the undivided cell population without any CFSE dye-dilution. For individual CFSE peaks, the total number of cell divisions were then calculated by multiplying the assigned division number with the number of starting cell in that peak. Finally, the division index was calculated by dividing the combined total cell division numbers of all peaks by the combined starting cell numbers of all peaks.
Cell sorting
For in vitro T cell proliferation and in vivo homeostatic expansion, T cells were purified to 88–95% purity via negative selection of B220+, CD11b+, CD49b+, and Ter119+ cells from pooled spleen and lymph node (LN) cells using biotin-conjugated antibodies (BioLegend), streptavidin-microbeads and MACS columns (Miltenyi Biotec, 130-048-101 and 130-042-401) with standard protocols. For qPCR analyses of ppp4c deletion efficiency, MACS-purified T cells were further sorted into CD4 and CD8 T cells by flow cytometry on FACSAria (BD Biosciences). Catalog numbers of the antibodies are available upon request.
Cell culture
For T cell proliferation, MACS-purified T cells from pooled spleen and LN were loaded with CFSE and cultured at 2 × 105 cells per well in 96-well plates with 1.6 μg/ml plate-bound anti-CD3ε and anti-CD28 (BD Biosciences). For the competitive proliferation assay, purified congenic-WT (CD90.1+/CD90.2−) and CD4cre:PP4f/f (CD90.1−/CD90.2+) T cells were mixed at equal ratios, loaded with CFSE and cultured as above. For cell cycle analysis, 10 μM BrdU was added to the culture media during the last 2 hr of incubation. For T cell proliferation with exogenous IL-2, purified T cells were activated as above, except using a lower dose of 0.5 μg/ml anti-CD3ε and anti-CD28, with or without 100 ng/mL exogenous IL-2. For TCR signaling, MACS-purified T cells were stimulated with 1 μg/ml biotin-labeled anti-CD3ε and anti-CD28 (BD Biosciences); this was then followed by crosslinking with 1 μg/ml streptavidin at 37°C for the indicated time. Primary T cells were cultured in DMEM supplemented with 1x non-essential amino acid, 2 mM L-glutamine, 2 mM Glutamax, 1 mM sodium pyruvate, 10 mM HEPES (Invitrogen), 10% FBS (BioLegend), 100 U/ml penicillin, 100 mg/ml streptomycin (Biological Industries), and 125 mM 2-ME (Sigma-Aldrich). Resveratrol and OA were purchased (Enzo Life Sciences, BML-FR104-0100 and ALX-350-003-C025). HEK-293 and the derivative clones were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin (Biological Industries). JTAg cells were cultured in RPMI-1640 supplemented with FBS and penicillin/streptomycin as above. Catalog numbers of the antibodies are available upon request.
Cytokine secretions
Culture supernatants from 3 μg/ml OVA re-stimulation, or those from anti-CD3ε- and anti-CD28-stimulated T cells were collected on day 3 and frozen until analyzed. Multiplex cytokine assay was performed with the Mouse Th1/Th2 10plex FlowCytomix kit (eBioscience, BMS820FFRTU) following the manufacturer's instructions, with the concentrations of secreted cytokines calculated using the titration curves of standards at a detection sensitivity of 10–20 pg/ml. All samples were measured in duplicate.
OVA/CFA immunization and nitrophenyl (NP)-specific antibody responses
For T cell re-stimulation following OVA/CFA immunization, WT and CD4cre:PP4f/f mice were immunized in both hind footpads with 0.1 ml of emulsion containing 1 mg/ml CFA (BD Biosciences/Difco, 263810) and 1 mg/ml OVA (Sigma-Aldrich, S7951-1MG). One week following immunization, T cells from the popliteal LN were harvested, loaded with CFSE, and cultured at 4 × 105 cells per well in 96-well U-bottom plates with titrating doses of OVA for 3 d prior to flow cytometry analyses. For primary and memory Ig responses, mice were immunized i.p. with 200 mg NP-keyhole limpet hemocyanin (KLH) (Biosearch Technologies, N-5060-25) emulsified with either 2 mg alum (Sigma-Aldrich, 255963) or 2 mg/ml CFA in a final volume of 200 μl, or with 100 μg/100 μl NP-Ficoll (Biosearch Technologies, F-1420-100), and subjected to sera collection and ELISA measurement to assess the production of NP-specific antibody. ELISA detection was performed using HRP-conjugated mouse isotype-specific secondary antibodies (Bethyl) and HRP substrate (BD Biosciences), with the results read on EnVision 2104 multi-label reader (Perkin-Elmer, Waltham). Catalog numbers of the antibodies are available upon request.
In vivo homeostasis expansion
MACS-purified CD90.1+/CD90.2− WT and CD90.1−/CD90.2+ CD4cre:PP4f/f T cells from pooled spleen and LNs of 4–6 wk old mice were mixed at a 1:1 ratio and labeled with CFSE as described above. RAG1−/− recipient mice were adoptively transferred with 2 ×106 cell mixtures via tail vein. At the specified time, recipient mice were sacrificed, and cells from their spleens and LNs were subjected to flow cytometry analyses.
Luciferase reporter assays
JTAg cells were transfected with the IL-2 promoter-LucCitation48 or the NFAT/AP1x3-Luc Citation49 luciferase reporters by Neon microporator (Invitrogen); tdTomato expression plasmid was co-transfected to serve as transfection efficiency control. One day after transfection, 50 ng/ml PMA (Sigma-Aldrich, P1585), 500 ng/ml ionomycin and various concentrations of OA were added to the culture for 6 hr. The luciferase activity was then measured using the Neolite luciferase reporter assay system (Perkin-Elmer, 6016711) on Luminometer TD 20/20 (Turner Designs), while a small fraction of the transfected cells was subjected to flow cytometry analyses for the percentages of tdTomato+ cells as an index of transfection efficiency.
Western blotting and co-immunoprecipitation analyses
Antibodies against phospho-4EBP1 (Thr-37/46), 4EBP1, phospho-AMPKα-subunit (Thr-172), phospho-Akt (Ser-473), phospho-CDK2 (Thr-160), phospho-ERK (Thr-202/Tyr-204), ERK, phospho-IKKα (Ser-180)/IKKβ (Ser-181), mTOR, phospho-mTOR (Thr-2448, Thr-2481), phospho-p38 (Thr-180/Tyr-182), phospho-S6K (Thr-389), S6K, and phospho-Zap-70 (Tyr-319)/Syk (Tyr-352) were purchased (Cell Signaling Technology). Antibodies against β-actin (Sigma-Aldrich), AMPKα (Millipore), CDK2, cyclin A, cyclin B1, cyclin D1, cyclin E, p27Kip1 (Santa Cruz Biotechnology), phospho-CDK2 (Thr-14) (Bioss), phospho-JNK (Tyr-185/Tyr-223) (Epitomics), KAP1 (Bethyl Laboratories), β-tubulin, Flag-tag and myc-tag (Genetex) were also purchased. Co-immunoprecipitation was performed with standard protocols using anti-Flag M2 affinity gel (Sigma-Aldrich, A2220) at 4°C on a rotating platform for 3 h. Lysate preparation and western blotting were performed with standard protocols and were visualized on BioSpectrum Imagining System with Vision-Works LS software (UVP). Catalog numbers of the antibodies are available upon request.
Generation of tetracycline-inducible HEK-293 clones
HEK-293 tetracycline-inducible cells were generated by first transfecting HEK-293 cells with pcDNA4-TR (Invitrogen) and selecting for blasticidin (4 μg/ml, Invitrogen)-resistant stable clones, and then subjected to a second transfection with pcDNA4-TO-PP4-Myc or pcDNA4-TO-GFP (Invitrogen), selection with Zeocin (400 μg/ml, Invitrogen), and limiting dilution sub-cloning. Tetracycline inducibility of the obtained clones was validated by protein gel blotting.
Lentivirus packaging and primary T cell infection
HEK-293T cells were transiently transfected with pCMV-ΔR8.91, pMD.G, and lentiviral vectors (National Core Facility for Manipulation of Gene Function by RNAi, miRNA, miRNA sponges, and CRISPR / Genomic Research Center) with standard protocols and cultured in 25 μM chloroquine over-night, and then subjected to chloroquine-free media change and additional culture for 24 hr. Lentiviral supernatants were then harvest 3 times with 24 hr intervals, pooled, and validated by serial dilution and infection. Lentiviral infection of CFSE-loaded primary T cells was performed by culturing cells with a 1:1 mixture of lentiviral supernatants and fresh culture media supplemented with 8 μg/ml polybrene at the time of stimulation by 1.6 μg/ml plate-bound anti-CD3ε and anti-CD28. Cells were harvested 48 hr later and analyzed by flow cytometry.
Statistical analyses
In all assays, data were plotted as mean ± SEM, with the p-values calculated by unpaired or paired 2-tailed Student's t-test.
Abbreviations
| 7AAD | = | 7-aminoactinomycin |
| ALN | = | auxiliary lymph node |
| AMPK | = | AMP-activated protein kinase |
| CA | = | constitutively-active |
| CD4-cre | = | CD4 promoter-driven Cre recombinase transgene |
| KLH | = | keyhole limpet hemocyanin |
| Lck-cre | = | proximal Lck promoter-driven Cre recombinase transgene |
| LN | = | lymph node |
| mTOR | = | mechanistic target of rapamycin |
| MLN | = | mesenteric lymph node |
| NHRI | = | National Health Research Institutes |
| NP | = | nitrophenyl |
| OA | = | okadaic acid |
| p- | = | phosphorylated |
| PP4 | = | protein phosphatase 4 |
| qPCR | = | quantitative PCR |
| Resv | = | resveratrol |
| ROS | = | reactive oxygen species |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2015CC6839R-f02-z-4c.pdf
Download PDF (475.2 KB)Acknowledgments
F. L. designed and performed experiments as well as analyzed data. W. H., Y. L. and Y. C. performed experiments. C. H. designed and performed experiments, analyzed data, and prepared the manuscript. We thank Dr. David McKean (via Addgene plasmid#11783) for the NFAT/AP1x3 luciferase reporter plasmid, Dr. Anjana Rao (via Addgene#12194) for the IL-2 promoter luciferase reporter plasmid, and Claire Y. Huang for proof-reading the manuscript. We also thank the staff of the Laboratory Animal Center and the sequencing/flow cytometry core facility at NHRI for their assistance.
Funding
This work was supported in whole or in part by grants 03-A1-IMPP03-014 (NHRI, Taiwan) and 101-2320-B-400-004-MY3/103-2321-B-400-004 (Ministry of Science and Technology, Taiwan) to C.H. F.L. was supported by a post-doctoral fellowship from the Ministry of Science and Technology, Taiwan.
References
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol 2009; 27:591-619; PMID:19132916; http://dx.doi.org/10.1146/annurev.immunol.021908.132706
- von Boehmer H. Selection of the T-cell repertoire: receptor-controlled checkpoints in T-cell development. Adv Immunol 2004; 84:201-38; PMID:15246254; http://dx.doi.org/10.1016/S0065-2776(04)84006-9
- Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013; 38:13-25; PMID:23352221; http://dx.doi.org/10.1016/j.immuni.2013.01.004.
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science 2013; 342:1242454; PMID:24115444; http://dx.doi.org/10.1126/science.1242454
- Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol 2012; 13:907-15; PMID:22990888; http://dx.doi.org/10.1038/ni.2386.
- Mills RE, Jameson JM. T cell dependence on mTOR signaling. Cell Cycle 2009; 8:545-8; PMID:19182526; http://dx.doi.org/10.4161/cc.8.4.7625
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 2009; 9:324-37; PMID:19390566; http://dx.doi.org/10.1038/nri2546
- Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med 2008; 14:539-49; PMID:18977694; http://dx.doi.org/10.1016/j.molmed.2008.09.007
- Auciello FR, Ross FA, Ikematsu N, Hardie DG. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett 2014; 588:3361-6; PMID:25084564; http://dx.doi.org/10.1016/j.febslet.2014.07.025
- Blagih J, Krawczyk CM, Jones RG. LKB1 and AMPK: central regulators of lymphocyte metabolism and function. Immunol Rev 2012; 249:59-71; PMID:22889215; http://dx.doi.org/10.1111/j.1600-065X.2012.01157.x
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res 2010; 70:1042-52; PMID:20103647; http://dx.doi.org/10.1158/0008-5472.CAN-09-3537
- Suzuki Y, Ito S, Sasaki R, Asahi M, Ishida Y. Resveratrol suppresses cell proliferation via inhibition of STAT3 phosphorylation and Mcl-1 and cIAP-2 expression in HTLV-1-infected T cells. Leuk Res 2013; 37:1674-9; PMID:24090995; http://dx.doi.org/10.1016/j.leukres.2013.09.010
- Zou T, Yang Y, Xia F, Huang A, Gao X, Fang D, Xiong S, Zhang J. Resveratrol Inhibits CD4+ T cell activation by enhancing the expression and activity of Sirt1. PLoS One 2013; 8:e75139; PMID:24073240; http://dx.doi.org/10.1371/journal.pone.0075139
- Shui JW, Hu MC, Tan TH. Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol Cell Biol 2007; 27:79-91; PMID:17060460; http://dx.doi.org/10.1128/MCB.00799-06
- Liao FH, Shui JW, Hsing EW, Hsiao WY, Lin YC, Chan YC, Tan TH, Huang CY. Protein phosphatase 4 is an essential positive regulator for Treg development, function, and protective gut immunity. Cell Biosci 2014; 4:25; PMID:24904742; http://dx.doi.org/10.1186/2045-3701-4-25
- Shaltiel IA, Aprelia M, Saurin AT, Chowdhury D, Kops GJ, Voest EE, Medema RH. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc Natl Acad Sci U S A 2014; 111:7313-8; PMID:24711418; http://dx.doi.org/10.1073/pnas.1322021111
- Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell 2008; 31:33-46; PMID:18614045; http://dx.doi.org/10.1016/j.molcel.2008.05.016
- Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep 2008; 9:1019-26; PMID:18758438; http://dx.doi.org/10.1038/embor.2008.162
- Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J 2012; 31:2403-15; PMID:22491012; http://dx.doi.org/10.1038/emboj.2012.86
- Lipinszki Z, Lefevre S, Savoian MS, Singleton MR, Glover DM, Przewloka MR. Centromeric binding and activity of Protein Phosphatase 4. Nat Commun 2015; 6:5894; PMID:25562660; http://dx.doi.org/10.1038/ncomms6894
- Sato-Carlton A, Li X, Crawley O, Testori S, Martinez-Perez E, Sugimoto A, Carlton PM. Protein phosphatase 4 promotes chromosome pairing and synapsis, and contributes to maintaining crossover competence with increasing age. PLoS Genet 2014; 10:e1004638; PMID:25340746; http://dx.doi.org/10.1371/journal.pgen.1004638
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol 2007; 365:23-38; PMID:17200551
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 2000; 80:173-210; PMID:10617768
- Richards FM, Milner J, Metcalfe S. Inhibition of the serine/threonine protein phosphatases PP1 and PP2A in lymphocytes: effect on mRNA levels for interleukin-2, IL-2R alpha, krox-24, p53, hsc70 and cyclophilin. Immunology 1992; 76:642-7; PMID:1328040
- Grove DS, Mastro AM. Modulation of levels of a negative transcription factor for IL-2 by 12-O-tetradecanoyl phorbol-13-acetate and okadaic acid. Cytokine 1996; 8:809-16; PMID:9047076; http://dx.doi.org/10.1006/cyto.1996.0108
- Samari HR, Moller MT, Holden L, Asmyhr T, Seglen PO. Stimulation of hepatocytic AMP-activated protein kinase by okadaic acid and other autophagy-suppressive toxins. Biochem J 2005; 386:237-44; PMID:15461583; http://dx.doi.org/10.1042/BJ20040609
- Lacroix I, Lipcey C, Imbert J, Kahn-Perles B. Sp1 transcriptional activity is up-regulated by phosphatase 2A in dividing T lymphocytes. J Biol Chem 2002; 277:9598-605; PMID:11779871; http://dx.doi.org/10.1074/jbc.M111444200
- Mourtada-Maarabouni M, Williams GT. Protein phosphatase 4 regulates apoptosis in leukemic and primary human T-cells. Leuk Res 2009; 33:1539-51; PMID:19539371; http://dx.doi.org/10.1016/j.leukres.2009.05.013
- Ohmura T, Onoue K. Stability of IL-2 mRNA in T lymphocytes is controlled by a protein kinase C-regulated mechanism. Int Immunol 1990; 2:1073-9; PMID:1964589; http://dx.doi.org/10.1093/intimm/2.11.1073
- Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J 1992; 11:3995-4005; PMID:1396589
- Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 2006; 203:1665-70; PMID:16818670; http://dx.doi.org/10.1084/jem.20052469
- Andris F, Leo O. AMPK in Lymphocyte Metabolism and Function. Int Rev Immunol 2014; PMID:25360847
- Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol 2014; 14:435-46; PMID:24962260; http://dx.doi.org/10.1038/nri3701
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283-93; PMID:15866171; http://dx.doi.org/10.1016/j.molcel.2005.03.027
- Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem 2007; 282:9777-88; PMID:17255104; http://dx.doi.org/10.1074/jbc.M608310200
- Moore F, Weekes J, Hardie DG. Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem 1991; 199:691-7; PMID:1678349; http://dx.doi.org/10.1111/j.1432-1033.1991.tb16172.x
- Wang L, Brautigan DL. alpha-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKalpha in vitro. Nat Commun 2013; 4:1559; PMID:23463002; http://dx.doi.org/10.1038/ncomms2565
- Liang X, Wang P, Gao Q, Tao X. Exogenous activation of LKB1/AMPK signaling induces G(1) arrest in cells with endogenous LKB1 expression. Mol Med Rep 2014; 9:1019-24; PMID:24469340
- Cai X, Hu X, Cai B, Wang Q, Li Y, Tan X, Hu H, Chen X, Huang J, Cheng J, et al. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle G1/G0 phase arrest and p21CIP and p27KIP expression and downregulation of cyclin D1 in vitro and in vivo. Oncol Rep 2013; 30:2449-57; PMID:24008375
- Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7:667-77; PMID:16921403; http://dx.doi.org/10.1038/nrm1987
- Valdor R, Macian F. Induction and stability of the anergic phenotype in T cells. Semin Immunol 2013; 25:313-20; PMID:24211041; http://dx.doi.org/10.1016/j.smim.2013.10.010
- Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol 2009; 183:6095-101; PMID:19841171; http://dx.doi.org/10.4049/jimmunol.0803510
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001; 15:763-74; PMID:11728338; http://dx.doi.org/10.1016/S1074-7613(01)00227-8
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992; 68:869-77; PMID:1547488; http://dx.doi.org/10.1016/0092-8674(92)90030-G
- He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011; 117:3869-80; PMID:21310926; http://dx.doi.org/10.1182/blood-2010-10-312736
- Hedin KE, Bell MP, Kalli KR, Huntoon CJ, Sharp BM, McKean DJ. Delta-opioid receptors expressed by Jurkat T cells enhance IL-2 secretion by increasing AP-1 complexes and activity of the NF-AT/AP-1-binding promoter element. J Immunol 1997; 159:5431-40; PMID:9548483
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J 2000; 19:4783-95; PMID:10970869; http://dx.doi.org/10.1093/emboj/19.17.4783