
ABSTRACT
Low-dose radiation risks remain unclear owing to a lack of sufficient studies. We previously reported that low-dose, long-term fractionated radiation (FR) with 0.01 or 0.05 Gy/fraction for 31 d inflicts oxidative stress in human fibroblasts due to excess levels of mitochondrial reactive oxygen species (ROS). To identify the small effects of low-dose radiation, we investigated how mitochondria respond to low-dose radiation in radiosensitive human ataxia telangiectasia mutated (ATM)- and Nijmegen breakage syndrome (NBS)1-deficient cell lines compared with corresponding cell lines expressing ATM and NBS1. Consistent with previous results in normal fibroblasts, low-dose, long-term FR increased mitochondrial mass and caused accumulation of mitochondrial ROS in ATM- and NBS1-complemented cell lines. Excess mitochondrial ROS resulted in mitochondrial damage that was in turn recognized by Parkin, leading to mitochondrial autophagy (mitophagy). In contrast, ATM- and NBS1-deficient cells showed defective induction of mitophagy after low-dose, long-term FR, leading to accumulation of abnormal mitochondria; this was determined by mitochondrial fragmentation and decreased mitochondrial membrane potential. Consequently, apoptosis was induced in ATM- and NBS1-deficient cells after low-dose, long-term FR. Antioxidant N-acetyl-L-cysteine was effective as a radioprotective agent against mitochondrial damage induced by low-dose, long-term FR among all cell lines, including radiosensitive cell lines. In conclusion, we demonstrated that mitochondria are target organelles of low-dose radiation. Mitochondrial response influences radiation sensitivity in human cells. Our findings provide new insights into cancer risk estimation associated with low-dose radiation exposure.
KEYWORDS:
Introduction
The effects of ionizing radiation (IR) have been well investigated using acute single radiation at high doses. Double-strand breaks (DSBs) in the cell nucleus produced by IR are considered to trigger genomic instability, a hallmark of cancer in irradiated cells. Mammalian cells harbor cellular defense systems against DSBs, including activation of cell cycle checkpoints, apoptosis, and DNA repair mechanisms, to maintain genome integrity.Citation1 Although health risks associated with low-dose radiation are currently being investigated intensively, the effects of low-dose radiation remain unclear owing to a lack of sufficient studies. Recently, we reported that low-dose, long-term fractionated radiation (FR) with 0.01 or 0.05 Gy/fraction induces oxidative stress in irradiated normal human fibroblasts via accumulation of mitochondrial reactive oxygen species (ROS).Citation2 Mitochondrial DNA is considered to be more susceptible to IR than nuclear DNA because mitochondrial DNA lacks histone protection and an efficient DNA repair system.Citation3 Thus, mitochondrial dysfunction can be used as a marker to assess the effect of low-dose radiation.
Mitochondria are ubiquitous intracellular organelles that generate adenosine triphosphate via oxidative phosphorylation. Mitochondria also regulate apoptosis via release of cytochrome c4 and play a crucial role in ROS generation as a byproduct of oxidative phosphorylation.Citation5 ROS function as second messengers for signal transduction and modulate various biological processes, including proliferation, differentiation, and metabolic adaptation to low-oxygen conditions.Citation5,6 However, ROS at higher levels inflict oxidative stress that induces DNA damage at a later time after IR. Such a delayed radiation effect may play a prominent role in IR-induced genomic instability.Citation7–10
A DNA damage sensor kinase, ataxia telangiectasia mutated (ATM), recognizes DNA DSBs and relays DNA damage signals to several target proteins. The MRE11-RAD50-NBS1 complex is recruited to DSBs for activating ATM and promotes DSB repair via various pathways, including homologous recombination repair.Citation11–13 The genetic disorders ataxia telangiectasia and Nijmegen breakage syndrome (NBS), which are characterized by high radiosensitivity, have mutations in the ATM gene and in the p95/nbs1 gene, respectively.Citation14,15 To identify the relatively small effects of low-dose radiation, we used highly radiosensitive human ATM- and NBS1-deficient cells (AT5BIVA and GM7166, respectively), which are defective in the DNA damage response.
In this study, human ATM- and NBS1-deficient cell lines and corresponding cell lines that expressed ATM and NBS1 were exposed to 0.01 or 0.05 Gy/fraction of FR for 31 d. Mitochondrial damage and oxidative stress were investigated in these cells. We found that mitochondria are target organelles for low-dose, long-term FR. Additionally, we found that the antioxidant N-acetyl-L-cysteine (NAC) is capable of mitigating mitochondrial damage and radiation toxicity induced by low-dose, long-term FR.
Results
NAC protects against low-dose radiation-related toxicity in radiosensitive ATM- and NBS1-deficient cells
AT5BIVA (ATM-deficient), AT5BIVA/ATM-wt (ATM-complemented), GM7166 (NBS1-deficient), and GM7166/NBS1-wt (NBS1-complemented) cells were exposed to 0.01- or 0.05-Gy fractions of x-rays for 31 d. Cells thus treated with FR were termed 31FR cells. The 31FR cells of all 4 cell lines showed decreased colony survival after repeated exposure to low-dose FR. Survival was affected in a dose-dependent manner (). Severe sensitivity to low-dose, long-term FR was observed in ATM- and NBS1-deficient cells () compared with corresponding ATM- and NBS1-complemented cells (). For all 4 cell lines, continuous treatment with the antioxidant NAC during low-dose, long-term FR exposure resulted in protection against FR-induced cell death ().
Figure 1. Survival after low-dose, long-term fractionated radiation (FR) in ATM- and Nijmegen breakage syndrome (NBS)1-deficient cells. Survival of ataxia telangiectasia mutated- and NBS1-deficient cells and the corresponding complemented cells after low-dose long-term FR with and without N-acetyl-L-cysteine (NAC) treatment. Asterisk indicates a significant decrease in surviving fraction compared with unirradiated control cells.
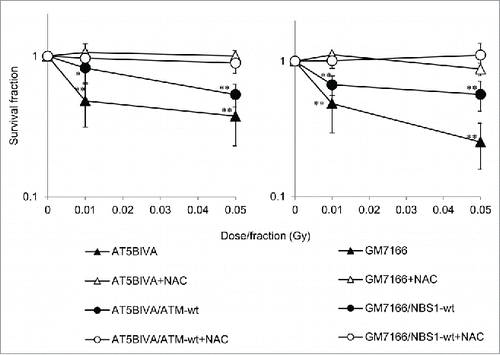
As we previously reported, low-dose, long-term FR induces growth retardation in all 4 cell lines used in this article.Citation16 However, continuous treatment with NAC during FR rescued cell proliferation in all 4 cell lines (Fig. S1). These results suggest that low-dose, long-term FR causes oxidative stress that results in decreased cell survival and delayed cell growth. NAC was shown to mitigate low-dose, long-term FR-related toxicity even in radiosensitive cell lines.
ROS induction after low-dose, long-term FR
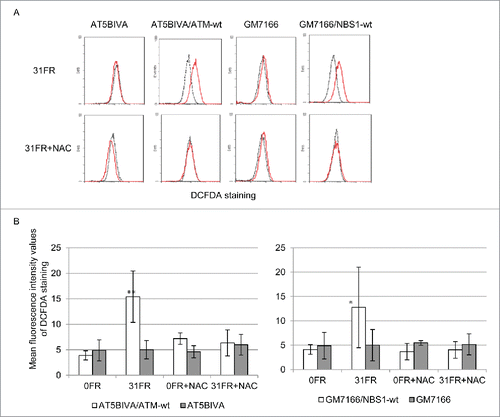
Oxidative stress triggered by low-dose, long-term FR may be caused by increased generation of ROS in irradiated cells. We used 2′,7′-dichlorofluorescin diacetate (DCFDA) staining to measure amounts of ROS in FR-treated cells (). As expected, ROS levels were increased in AT5VIBA/ATM-wt 31FR and GM7166/NBS1-wt 31FR cells compared with unirradiated 0FR cells (). NAC treatment counteracted the increased generation of ROS in 31FR cells (). In contrast, ROS levels were unaffected by low-dose, long-term FR in AT5BIVA 31FR and GM7166 31FR cells regardless of NAC treatment ().
Figure 2. Decreased levels of reactive oxygen species induced by low-dose long term fractionated radiation (FR) in ataxia telangiectasia mutated- and Nijmegen breakage syndrome 1-deficient cells. (A) Fluorescence-activated cell sorting results for 2′,7′-dichlorofluorescein diacetate (DCFDA) staining in unirradiated (0FR; dotted black lines) and cells irradiated for 31 d (31FR; red lines) with and without N-acetyl-L-cysteine. (B) Mean fluorescence intensity values of DCFDA staining. Asterisks indicate a significant increase in DCFDA staining compared with control 0FR cells.
Mitochondrial fragmentation in ATM- and NBS1-deficient cells after low-dose, long-term FR
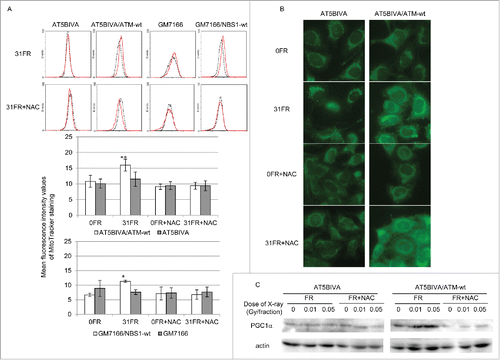
We next investigated the role of mitochondria in radiation response. Mitochondrial mass was measured using Mitotracker Green FM staining. Consistent with previous results in normal human fibroblasts,Citation2 the intensity of Mitotracker Green FM staining was increased in AT5BIVA/ATM-wt 31FR and GM7166/NBS1-wt 31FR cells compared with unirradiated cells (). It has been reported that ATM is required for IR-induced mitochondrial biogenesis via AMP-activated protein kinase activation.Citation17 Consistent with this, mitochondrial mass was unchanged in ATM- and NBS1-deficient 31FR cells (). Morphological observation of mitochondria indicated that mitochondrial fragmentation consistent with apoptosis was induced by low-dose, long-term FR in ATM- and NBS1-deficient 31FR cells ( and S2). NAC treatment prevents low-dose, long-term FR-induced mitochondrial fragmentation in ATM- and NBS1-deficient 31FR cells (). Expression of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), a marker for mitochondrial biogenesis, increased after low-dose, long-term FR in ATM-complemented cells but not in ATM-deficient 31FR cells. NAC treatment prevented the increase in mitochondrial mass and induction of PGC1α expression caused by low-dose, long-term FR in ATM-complemented 31FR cells ().
Figure 3. Mitochondrial mass after fractionated radiation (FR). (A) Fluorescence-activated cell sorting results for MitoTracker Green FM staining in unirradiated cells (0FR; dotted black lines) and cells irradiated for 31 d (31FR; red lines) in ataxia telangiectasia mutated (ATM)- and Nijmegen breakage syndrome 1-deficient cell 1-deficient cells and the corresponding complemented cell lines. Mean fluorescence intensity values of MitoTracker staining are shown in the lower panel. Asterisks indicate a significant increase in mitochondrial mass compared with control 0FR cells. (B) Images of MitoTracker Green FM staining in 0FR and 31FR cells of ATM-deficient cells and complemented cells with and without N-acetyl-L-cysteine. (C) Western blotting for peroxisome proliferator-activated receptor-γ coactivator 1α and actin in 0FR and 31FR cells with and without NAC.
Lack of induction of mitophagy after low-dose, long-term FR in ATM- and NBS1-deficient cells
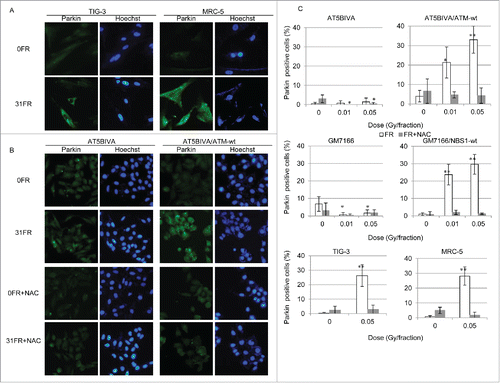
ROS are highly reactive molecules that can oxidize biomolecules. Therefore, mitochondrial ROS may cause mitochondrial damage in 31FR cells. We examined immunostaining for the E3 ubiquitin ligase Parkin, which marks damaged mitochondria for degradation via mitophagy. Parkin-positive cells were induced by low-dose, long-term FR in MRC-5 and TIG-3 31FR cells as well as in ATM- and NBS1-complemented 31FR cells (). NAC treatment suppressed staining for Parkin in ATM- and NBS1-complemented 31FR cells (). In contrast, Parkin staining was negative in ATM- and NBS1-deficient 31FR cells regardless of NAC treatment (). These results suggest that ATM and NBS1 are required for localization of Parkin in damaged mitochondria after low-dose, long-term FR.
Figure 4. Immunofluorescent staining for Parkin after fractionated radiation (FR). (A) IF localization of Parkin in unirradiated TIG-3 and MRC-5 cells (0FR) and cells irradiated for 31 d (31FR). DNA was stained with Hoechst. (B) Images of Parkin staining in 0FR and 31FR ataxia telangiectasia mutated-deficient cells and complemented cells with and without N-acetyl-L-cysteine (NAC) treatment. (C) Percentage of Parkin-positive cells in indicated cells exposed to 0.01- or 0.05-Gy fractions and exposed to FR plus NAC. Asterisks indicate a significant increase or decrease in the percentage of Parkin-positive cells compared with control 0FR cells.
Decrease in mitochondrial potential and induction of apoptosis in ATM- and NBS1-deficient 31FR cells
Damaged mitochondria may accumulate after low-dose, long-term FR in ATM- and NBS1-deficient cells owing to defective mitophagy. To investigate abnormalities in mitochondria, mitochondrial membrane potential (Δψm) was measured by staining with the lipophilic cation JC-1 probe. A decrease in Δψm was evident in ATM-deficient 31FR cells, as shown by negative staining for JC-1 (). In contrast, mitochondrial membrane potential was unaffected by low-dose, long-term FR in ATM-complemented 31FR cells as shown by positive staining for JC-1 ().
Figure 5. Mitochondrial membrane potential and apoptosis in ataxia telangiectasia mutated (ATM)- and Nijmegen breakage syndrome 1-deficient cell (NBS)1-deficient cells after fractionated radiation (FR). (A) Images of JC-1 staining in unirradiated (0FR) and 31-day irradiated (31FR) ATM-deficient and -complemented cells. (B) Annexin V staining in 0FR and 31FR cells with and without N-acetyl-L-cysteine (NAC). (C) Percentage of apoptotic cells in ATM- and NBS1-deficient 31FR cells and NBS1-complemented 31FR cells with NAC. Asterisks indicate a significant increase in the incidence of apoptosis compared with control 0FR cells.
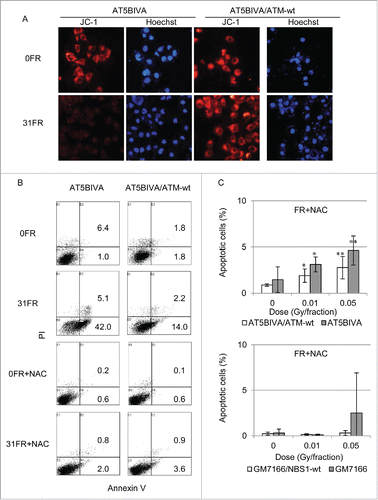
We next examined the incidence of apoptosis after low-dose, long-term FR (). Apoptosis was examined using an apoptosis detection kit in ATM- or NBS1-complemented cells and ATM- or NBS1-deficient cells. We previously reported that low-dose, long-term FR induced apoptosis in all 4 cell lines.Citation16 As assessed by staining for annexin V, 47.1% of ATM-deficient 31FR cells were apoptotic, whereas 16.2% of ATM-complemented 31FR cells were apoptotic (). NAC treatment suppressed apoptosis (less than 5%) in all 31FR cells ().
Nuclear DNA damage in ATM- and NBS1-deficient 31FR cells
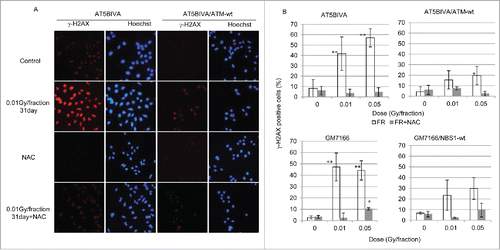
We monitored DSBs in nuclei after low-dose, long-term FR using the marker γ-H2AX (). γ-H2AX foci were induced after low-dose FR for >7 d in both ATM-complemented and ATM-deficient cells (data not shown). The percentage of γ-H2AX-positive cells was approximately 20% in ATM-complemented 31FR cells but was 40% or more in ATM-deficient 31FR cells (). Similarly, an increase in γ-H2AX-positive cells was more evident in NBS1-deficient cells compared with NBS1-complemented cells (). Administration of NAC prevented induction of γ-H2AX foci after low-dose, long-term FR in all 4 cell lines ().
Figure 6. γ-H2AX foci formation after fractionated radiation (FR). (A) Images of γ-H2AX-positive cells (red). DNA was stained with Hoechst. (B) The percentage of cells with γ-H2AX foci is shown for ataxia telangiectasia mutated-deficient and -complemented cells (left panel). Asterisks indicate a significant increase in γ-H2AX-positive cells compared with control unirradiated (0FR) cells.
Discussion
Mitochondrial dysfunction after low-dose, long-term FR
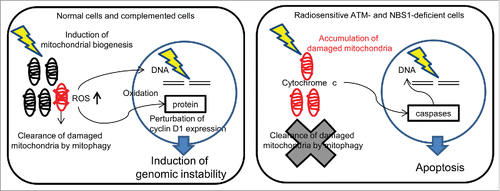
We investigated the effect of low-dose radiation on mitochondria in human cells. depicts differences in mitochondrial sensitivity to low-dose radiation between normal cells and radiosensitive ATM- and NBS1-deficient cells. Several reports have described increased mitochondrial DNA copy number after IR in vitro and in vivo.Citation3 IR stimulates mitochondrial biogenesis, mitochondrial gene expression, and mitochondrial activity to increase energy supply for radiation response.Citation3,18,19 Increase in mitochondrial mass and DNA copy number results in an overproduction of mitochondrial protein and alteration of electron transport chain activity; this in turn leads to an elevated superoxide production. Mitochondria express the antioxidant glutathione (GSH) to protect cells against oxygen toxicity.Citation20 However, low-dose, long-term FR decreases cellular GSH levels by GSH consumption. Insufficient GSH levels lead to a decrease in antioxidant capacity, persistent mitochondrial ROS accumulation, and long-lasting oxidative stress stimulation in long-term FR cells.Citation2 We demonstrated that repeated low-dose radiation induced mitochondrial biogenesis with elevated ROS levels in ATM- and NBS1-complemented cells (). As sites of ROS production via the electron transport chain, mitochondria are particularly vulnerable to ROS-induced damage. The protein Parkin recognizes damaged mitochondria and promotes their clearance by mitophagy.Citation21,22 Our present data show that after low-dose, long-term FR of normal human fibroblasts and ATM-and NBS1-complemented cells, Parkin accumulated in damaged mitochondria to control mitochondrial quality via induction of mitophagy. In contrast, mitochondrial biogenesis was not triggered by the same radiation treatments in ATM- and NBS1-deficient cells. In these cells, mitophagy was also defective, leading to accumulation of damaged mitochondria and, eventually, apoptosis ().
Figure 7. Schematic representation of mitochondrial damage induced by low-dose, long-term fractionated radiation and double-strand break repair in both ataxia telangiectasia mutated- and Nijmegen breakage syndrome 1-complemented and -deficient cells.
The role of ATM in response to low-dose, long-term FR
ATM has a role as a redox sensor, and under oxidative stress, it is activated by oxidization at a cysteine residue independent of DSBs.Citation23,24 ATM is required for IR-induced mitochondrial biogenesis.Citation17 Thus, an increase in mitochondrial mass was not observed after low-dose, long-term FR in ATM-deficient 31FR cells in contrast to ATM-complemented 31FR cells. ATM has also been shown to be associated with induction of mitophagy in response to IR and oxidative stress.Citation25 ATM loss leads to decreased mitophagy, mitochondrial dysfunction, and persistent oxidative stress in ATM-null mice.Citation25 In ATM-deficient 31FR cells, we found defective mitophagy, which led to accumulation of abnormal mitochondria with low Δψm. The permeabilization of the mitochondrial outer membrane results in the release of proapoptotic proteins such as cytochrome c and apoptosis-inducing factor to facilitate the activation of specific caspases and initiate a cascade of protease activation events (). Consequently, mitochondria-mediated apoptosis in ATM-deficient cells after low-dose, long-term FR leads to a highly radiosensitive phenotype with mitochondria-mediated apoptosis and severe growth retardation.
Mitochondria as target organelles for low-dose radiation and antioxidants as radioprotective agents against mitochondrial damage
We demonstrated that low-dose radiation induced mitochondrial ROS-mediated oxidative stress in complemented cells expressing ATM and NBS1, whereas it caused severe mitochondrial damage in radiosensitive cell lines. Thus, the radiation response of mitochondria influenced cell fate after IR. Mitochondrial dysfunction can be communicated to the cell nucleus via mitochondrial ROS acting as signaling molecules. Damage to nuclear DNA was evident long after low-dose FR by the persistence of γ-H2AX, a marker of DSBs. Mitochondrial DNA mutations by ROS-mediated oxidative modifications lead to progressive electron transport chain dysfunction and to further increases in ROS production, establishing a vicious cycle of mitochondrial ROS production.Citation26–28 If oxidative stress persists for prolonged periods, oxidative damage will accumulate in biomolecules and then induce mutagenesis, carcinogenesis, accelerated senescence, and cell death. We previously reported that mitochondrial ROS disrupt AKT/cyclin D1 cell cycle signaling via oxidative inactivation of protein phosphatase 2A, which is a negative regulator of AKT activity.Citation2 Resulting cyclin D1 nuclear accumulation is associated with cellular senescence and induction of genomic instability in irradiated cells ().Citation29–32 Thus, the effect of low-dose, long-term FR persists long after IR via oxidative stress triggered by chronically high levels of mitochondrial ROS. Collectively, mitochondrial dysfunction and subsequently elevated levels of ROS are implicated in the radiation-induced genomic instability of irradiated cells. NAC serves as cysteine donor for the synthesis of GSH and increases intracellular levels of GSHCitation33 for the suppression of accumulation of mitochondrial ROS. Data from our current study indicate that NAC suppressed low-dose FR-induced mitochondrial damage in all 4 cell lines studied, including the radiosensitive cell lines. Thus, increasing antioxidant capacity is critical to preventing radiation toxicity induced by low-dose, long-term FR.
In conclusion, we demonstrated that low-dose, long-term FR targets mitochondrial function. Excess mitochondrial ROS induced oxidative stress in normal cells, whereas apoptosis was induced in radiosensitive cells. Therefore, antioxidants may be useful agents for radioprotection against mitochondrial damage induced by low-dose, long-term FR.
Materials and methods
Cell culture conditions and drugs
ATM-defective human fibroblasts (AT5BIVA), ATM-wt reconstituted cells (AT5BIVA/ATM-wt), NBS1-defective human fibroblasts (GM7166), and NBS1-wt reconstituted cells (GM7166/NBS1-wt) were obtained from the Radiation Biology Center of Kyoto University. These cells were transformed with SV-40 and grown in RPMI 1640 medium (Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated fetal calf serum. Normal human diploid lung fibroblasts (MRC-5 and TIG-3) were purchased from the Health Science Research Resources Bank (Osaka, Japan) and grown in minimum essential medium (Nacalai Tesque) supplemented with 10% heat-inactivated fetal calf serum. NAC was purchased from Sigma (San Diego, CA, USA) and dissolved in water. Cells were continuously treated with NAC at a final concentration of 1 mM. Medium changes were made at 2- or 3-day intervals. Untreated cells were used as controls.
Irradiation experiments
Cells were irradiated using a 150-kVp X-ray generator (Model MBR-1505R2; Hitachi, Japan) with a 0.5-mm Cu and 0.1-mm Al filter at a dose of 0.7 Gy/min. Fractionation schedules were conducted as described previously.Citation30 Total doses delivered over 31 d were 0.46 and 2.3 Gy for cells exposed to FR of 0.01 and 0.05 Gy, respectively.
Clonogenic assay
Clonogenic survival of cells was assayed by seeding 103 cells per 60-mm dish. Dishes were incubated for 7—10 d until colonies were visible. Cells were fixed with ethanol for 30 min and stained with Giemsa solution (Merck, Pennsylvania, PA, USA). Colonies of more than 50 cells were counted under a light microscope.
Cell growth assay
Cells (5 × 105) were seeded into 25-cm2 flasks (Thermo Fisher Scientific), incubated overnight, and irradiated daily. Growth rates were monitored by counting cell numbers twice a week. When the total cell number exceeded 5 × 105, cells were subcultured to 5 × 105 cells in a new flask.
ROS detection and mitochondrial mass measurement
Cells were stained with 20 μM DCFDA (Sigma) or 400 nM MitoTracker Green FM (Invitrogen, Carlsbad, CA, USA) in minimum essential medium without serum for 30 min at 24 h after the last FR on the indicated days. DCFDA- or MitoTracker Green FM-stained cells were quantified with a FACScan (Becton Dickinson, USA). Cells were placed on glass slides and cultured overnight. Cells on coverslips were stained with MitoSOX-red or MitoTracker Green FM according to the manufacturer's instructions (Invitrogen). Images were acquired using a CCD camera attached to a fluorescence microscope (Keyence, Osaka, Japan).
Western blot analyses
Western blotting was performed as described previously.Citation34 The protein bands were visualized with Chemi-Lumi One L Western blotting substrate (Nacalai Tesque). Band intensity was measured by densitometry using Image Lab software (Bio-Rad).
Immunofluorescence staining
Immunofluorescence (IF) staining was performed as described previously.Citation34 Images were captured with a CCD camera attached to a fluorescence microscope (Keyence). To quantify IF data, more than 100 cells were counted per sample, with at least 3 independent samples.
Mitochondrial membrane potential
Cells on coverslips were stained with JC-1 according to the manufacturer's instructions (Invitrogen). Cells were incubated in JC-1 for 15 min. Images were acquired using a CCD camera attached to a fluorescence microscope (Keyence).
Annexin V staining
Apoptotic cells were identified and quantified using an annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (Bio Vision, Mountain View, CA, USA) following the manufacturer's protocol. Cells were stained with annexin V-FITC and propidium iodide 48 h after treatment with radiation. Annexin V-positive apoptotic cells were analyzed using fluorescence-activated cell sorting (FACScan; Becton Dickinson).
Statistical analysis
Error bars represent standard deviations; in some cases, the standard deviations were too small to be visible on the histogram. All the experiments were repeated at least 3 times with independent samples. One-way analysis of variance was used to determine whether there were any significant differences between the means of 3 or more independent groups. Single and double asterisks indicate significance of p < 0.05 and p < 0.01, respectively.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2016CC6991R1-s02.pptx
Download MS Power Point (968.4 KB)Acknowledgments
The authors thank the staff of the National Institute of Public Health for assistance with the research. We thank ScienceDocs Inc. (https://www.sciencedocs.com) and Enago (www.enago.jp) for language editing.
Funding
This research was supported by a grant from the Japanese Ministry of Education and Science Houga (15K12220), Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labor, and Welfare and in part by NIFS Collaborative Research Program (NIFS13KOBA028). This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University.
References
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433-9; PMID:11100718; http://dx.doi.org/10.1038/35044005.
- Shimura T, Sasatani M, Kamiya K, Kawai H, Inaba Y, Kunugita N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in low-dose irradiated human fibroblasts. Oncotarget 2015.
- Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med 2013; 65:607-19; PMID:23892359; http://dx.doi.org/10.1016/j.freeradbiomed.2013.07.024.
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996; 86:147-57; PMID:8689682; http://dx.doi.org/10.1016/S0092-8674(00)80085-9.
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48:158-67; PMID:23102266; http://dx.doi.org/10.1016/j.molcel.2012.09.025.
- Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J 1997; 11:118-24; PMID:9039953.
- Limoli CL, Giedzinski E, Morgan WF, Swarts SG, Jones GD, Hyun W. Persistent oxidative stress in chromosomally unstable cells. Cancer Res 2003; 63:3107-11; PMID:12810636.
- Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis 1996; 17:1633-9; PMID:8761419; http://dx.doi.org/10.1093/carcin/17.8.1633.
- Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis 2006; 21:361-7; PMID:17065161; http://dx.doi.org/10.1093/mutage/gel048.
- Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res 2006; 66:10377-83; PMID:17079457; http://dx.doi.org/10.1158/0008-5472.CAN-05-3036.
- Bruhn C, Zhou ZW, Ai H, Wang ZQ. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep 2014; 6:182-95; PMID:24388752; http://dx.doi.org/10.1016/j.celrep.2013.12.018.
- Carr AM, Lambert S. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 2013; 425:4733-44; PMID:23643490; http://dx.doi.org/10.1016/j.jmb.2013.04.023.
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308:551-4; PMID:15790808; http://dx.doi.org/10.1126/science.1108297.
- McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep 2004; 5:772-6; PMID:15289825; http://dx.doi.org/10.1038/sj.embor.7400210.
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; http://dx.doi.org/10.1038/nrm3546.
- Shimura T, Kobayashi J, Komatsu K, Kunugita N. DNA damage signaling guards against perturbation of cyclin D1 expression triggered by low-dose long-term fractionated radiation. Oncogenesis 2014; 3:e132; PMID:25486524; http://dx.doi.org/10.1038/oncsis.2014.48.
- Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PloS One 2008; 3:e2009; PMID:18431490; http://dx.doi.org/10.1371/journal.pone.0002009.
- Yu J, Wang Q, Chen N, Sun Y, Wang X, Wu L, et al. Mitochondrial transcription factor A regulated ionizing radiation-induced mitochondrial biogenesis in human lung adenocarcinoma A549 cells. J Radiat Res 2013; 54:998-1004; PMID:23645454; http://dx.doi.org/10.1093/jrr/rrt046.
- Kulkarni R, Marples B, Balasubramaniam M, Thomas RA, Tucker JD. Mitochondrial gene expression changes in normal and mitochondrial mutant cells after exposure to ionizing radiation. Radiat Res 2010; 173:635-44; PMID:20426663; http://dx.doi.org/10.1667/RR1737.1.
- Meister A. Glutathione, ascorbate, and cellular protection. Cancer Res 1994; 54:1969s-75s; PMID:8137322.
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 2003; 100:4078-83; PMID:12642658; http://dx.doi.org/10.1073/pnas.0737556100.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; http://dx.doi.org/10.1083/jcb.200809125.
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517-21; PMID:20966255; http://dx.doi.org/10.1126/science.1192912.
- Guo Z, Deshpande R, Paull TT. ATM activation in the presence of oxidative stress. Cell Cycle 2010; 9:4805-11; PMID:21150274; http://dx.doi.org/10.4161/cc.9.24.14323.
- Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012; 119:1490-500; PMID:22144182; http://dx.doi.org/10.1182/blood-2011-08-373639.
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014; 94:909-50; PMID:24987008; http://dx.doi.org/10.1152/physrev.00026.2013.
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006; 1757:509-17; PMID:16829228; http://dx.doi.org/10.1016/j.bbabio.2006.04.029.
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192:1001-14; PMID:11015441; http://dx.doi.org/10.1084/jem.192.7.1001.
- Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle 2013; 12:773-82; PMID:23388457; http://dx.doi.org/10.4161/cc.23719.
- Shimura T, Hamada N, Sasatani M, Kamiya K, Kunugita N. Nuclear accumulation of cyclin D1 following long-term fractionated exposures to low-dose ionizing radiation in normal human diploid cells. Cell Cycle 2014; 13:1248-55; PMID:24583467; http://dx.doi.org/10.4161/cc.28139.
- Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-9; PMID:23187803; http://dx.doi.org/10.4161/cc.22937.
- Dulic V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A 1993; 90:11034-8; PMID:8248208; http://dx.doi.org/10.1073/pnas.90.23.11034.
- Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A 1990; 87:9943-7; PMID:2263644; http://dx.doi.org/10.1073/pnas.87.24.9943.
- Shimura T, Toyoshima M, Adiga SK, Kunoh T, Nagai H, Shimizu N, Inoue M, Niwa O. Suppression of replication fork progression in low-dose-specific p53-dependent S-phase DNA damage checkpoint. Oncogene 2006; 25:5921-32; PMID:16682953; http://dx.doi.org/10.1038/sj.onc.1209624.