
Normal differentiated cells under normoxic conditions rely primarily on mitochondrial oxidative phosphorylation to generate the energy needed for cellular processes by oxidizing glucose-derived pyruvate in the mitochondrial tricarboxylic acid cycle to carbon dioxide. In contrast, most tumor cells exhibit elevated glucose uptake and lactate production regardless of oxygen levels with reduced mitochondrial oxidative phosphorylation.Citation1 This phenomenon, known as aerobic glycolysis or the Warburg effect, allows tumor cells to use a large fraction of glucose metabolites for synthesising macromolecules, such as amino acids, phospholipids, and nucleic acids, to facilitate tumor cell growth.Citation1
The Warburg effect is promoted by oncogenic signals such as those induced by activation of growth factor receptors including frequently overexpressed or mutated epidermal growth factor receptor (EGFR) in cancer.Citation2 EGF stimulation that activates Ras results in ERK1/2-mediated phosphorylation of S37 of pyruvate kinase M2 (PKM2), a rate-limiting glycolytic enzyme, which catalyzes the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP.Citation3 ERK1/2-phosphorylated PKM2 recruits peptidyl-proline isomerase protein interacting with never in mitosis A 1 (PIN1) for cis-trans isomerization of PKM2, which exposes the nuclear localization signal of PKM2 for binding to importin α5 and subsequent nuclear translocation.Citation3 In the nucleus, PKM2 binds to c-Src-phosphorylated β-catenin and functions as a protein kinase to phosphorylate histone H3 at T11, which leads to HDAC3 removal from the gene promoter regions to enhance the expression of CCND1 (encoding for cyclin D1) and MYC.Citation2,4 Cyclin D1 expression accelerates G1–S transition while c-Myc expression promotes the expression of glycolytic enzyme genes, including PKM2, GLUT1, and LDHA, thereby promoting the Warburg effect in a nuclear PKM2-depedent feedback loop.Citation5 In addition to PKM2-depdendent regulation of nuclear β-catenin, ERK1/2 phosphorylates and activates CK2α, leading to CK2α-dependent α-catenin S641 phosphorylation and subsequent α-catenin dissociation from β-catenin in adherence junctions for enhanced nuclear translocation and transactivation of β-catenin.Citation6 Thus, EGFR-induced activation of Ras and ERK is important for nuclear translocation of PKM2 and β-catenin transactivation–dependent expression of c-Myc, cyclin D1, and PKM2 to promote the Warburg effect and cell cycle progression.
Hyperactivation of the Ras, which results from growth factor receptor activation or Ras mutation, plays a critical role for tumorigenesis. The Ras proteins are active in guanosine triphosphate (GTP)-bound form, which can be regulated by guanine exchange factors (GEF), such as Son of Sevenless (SOS), and inactive in guanosine diphosphate (GDP)-bound form, which can be regulated by Ras GTPase-activating proteins (GAPs), such as p120-RasGAP and neurofibromin 1.Citation7
Our recent studies have uncovered an instrumental role of family with sequence similarity 129, member B (FAM129B, also known as MINERVA) in activation of Ras.Citation7 FAM129B is up-regulated in many types of cancer, including breast, kidney, large intestine, lung, endometrial cancers, and haematopoietic and central nervous system tumors. However, the mechanism by which FAM129B is regulated, thereby promoting tumor development, was unclear. We showed that EGFR activation results in increased binding of SOS to H-Ras while p120-RasGAP was dissociated from H-Ras upon EGF stimulation. Depletion of FAM129B, which did not affect the binding of SOS to Ras and activity of SOS, increased the basal level of the interaction between p120-RasGAP and H-Ras and largely abrogated EGF-induced dissociation of p120-RasGAP from H-Ras, thereby inhibiting Ras activation. To understand the mechanism underlying this observation, we found that EGF stimulation induced a direct interaction between EGFR and FAM129B, leading to EGFR-dependent FAM129B phosphorylation at Y593. This phosphorylation allowed FAM129B to bind to both H-Ras and K-Ras, which facilitated the dissociation of p120RasGAP from Ras without affecting the binding of SOS to Ras. Consequently, FAM129B Y593 phosphorylation promoted a full activation of H-Ras and K-Ras and its downstream ERK1/2, which in turn activated β-catenin-dependent gene transcription by CK2α-mediated release of β-catenin from α-catenin complex and the interaction between nuclear PKM2 and β-catenin. Thus, FAM129B positively regulates β-catenin/TCF/LEF transcription complex promoted cyclin D1, c-Myc, and PKM2 expressions, which in turn promoted the cell cycle progression, the Warburg effect, tumor cell proliferation, and brain tumorigenesis in mice ().Citation7
Figure 1. FAM129B promotes Ras activation by dissociation of p120-RasGAP from Ras. EGFR phosphorylates FAM129B, resulting in binding of phosphorylated FAM129B to Ras and reduced the association of p120-RasGAP with Ras. Activated Ras enhanced ERK1/2-dependent β-catenin transactivation for the Warburg effect, tumor cell proliferation, and brain tumorigenesis.
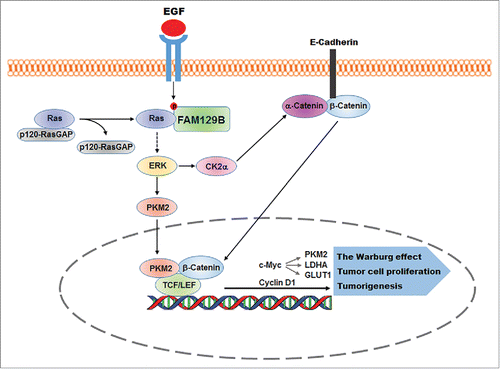
Given that FAM129B has been found highly expressed in many types of cancers, the identification of the essential function of FAM129B in activation of Ras and subsequent regulation of the Warburg effect and tumor cell proliferation provides new approaches to disrupt the oncogenic signaling from growth factor receptors and Ras for cancer treatment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Yang W, Lu Z. Cell Cycle 2013; 12:3154–8; PMID:24013426; http://dx.doi.org/10.4161/cc.26182
- Yang W,et al.Nature 2011; 480:118–22; PMID:22056988; http://dx.doi.org/10.1038/nature10598
- Yang W, et al.Nat Cell Biol 2012; 14:1295–304; PMID:23178880; http://dx.doi.org/10.1038/ncb2629
- Yang W, et al.Cell 2012; 150:685–96; PMID:22901803; http://dx.doi.org/10.1016/j.cell.2012.07.018
- Lu Z. Cell Cycle 2012; 11:4101–2; PMID:23070542; http://dx.doi.org/10.4161/cc.22325
- Ji H, et al.Mol Cell 2009; 36:547–59; PMID:19941816; http://dx.doi.org/10.1016/j.molcel.2009.09.034
- Ji H, et al.Proc Natl Acad Sci U S A 2016; 113:644–9; PMID:26721396; http://dx.doi.org/10.1073/pnas.1517112113