
ABSTRACT
Estrogen receptor alpha (ERα) has been implicated in several cell cycle regulatory events and is an important predictive marker of disease outcome in breast cancer patients. Here, we aimed to elucidate the mechanism through which ERα influences proliferation in breast cancer cells. Our results show that ERα protein is cell cycle-regulated in human breast cancer cells and that the presence of 17-β-estradiol (E2) in the culture medium shortened the cell cycle significantly (by 4.5 hours, P < 0.05) compared with unliganded conditions. The alterations in cell cycle duration were observed in the S and G2/M phases, whereas the G1 phase was indistinguishable under liganded and unliganded conditions. In addition, ERα knockdown in MCF-7 cells accelerated mitotic exit, whereas transfection of ERα-negative MDA-MB-231 cells with exogenous ERα significantly shortened the S and G2/M phases (by 9.1 hours, P < 0.05) compared with parental cells. Finally, treatment of MCF-7 cells with antiestrogens revealed that tamoxifen yields a slower cell cycle progression through the S and G2/M phases than fulvestrant does, presumably because of the destabilizing effect of fulvestrant on ERα protein. Together, these results show that ERα modulates breast cancer cell proliferation by regulating events during the S and G2/M phases of the cell cycle in a ligand-dependent fashion. These results provide the rationale for an effective treatment strategy that includes a cell cycle inhibitor in combination with a drug that lowers estrogen levels, such as an aromatase inhibitor, and an antiestrogen that does not result in the degradation of ERα, such as tamoxifen.
Introduction
Human estrogen receptor (ER) protein is a hormone-activated transcription factor, of which 2 forms have been identified to date: ERα and ERβ.Citation1 The transcription factor function of ERα depends on the association with its ligand 17-β-estradiol (E2). Upon ligand binding, ERα forms homodimers and translocates to the nucleus, where it can bind to estrogen response elements (EREs) of ERα target genes and initiate target gene transcription.Citation2 ERα can also act through ligand-independent mechanisms, including the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K) pathways.Citation3,4
Of the 5 molecular subtypes of breast tumors, the luminal A and B subtypes are ERα -positive tumors, which are associated with a better overall prognosis than human epidermal growth factor receptor 2 (HER2)-overexpressing tumors or ERα -negative, progesterone receptor (PR)-negative, and HER2-negative (triple-negative) tumors.Citation5,6 Among the ERα -positive subtypes, the clinical outcome of patients varies considerably, depending on the expression of the ESR1 gene, which encodes the ERα protein; luminal A tumors are correlated with higher levels of ESR1 expression and a better prognosis than are luminal B tumors.Citation6 ERα expression also plays an essential role in response to endocrine therapeutic agents, such as tamoxifen or fulvestrant. Tamoxifen is a selective estrogen receptor modulators (SERM) and exerts its antiestrogen functions by binding to ERα and inhibiting the transcription of estrogen-sensitive genes. Fulvestrant, on the other hand, is a selective estrogen receptor downregulator (SERD) that downregulates ERα, and, unlike tamoxifen, has no agonist activities on ERα. While tamoxifen increases the stability of ERα protein,Citation7,8 the binding of fulvestrant to ERα increases the rate of ERα protein degradation, effectively reducing the amount of cellular ERα protein.Citation9,10
One of the hallmarks of cancer is uncontrolled cell division, and deregulated expression of key cell cycle regulators, such as cyclins and cyclin-dependent kinases (CDKs), can trigger a cascade of events leading to mammary tumorigenesis. ERα has been implicated in several cell cycle regulatory events, including interactions with cyclins A and D1, which regulate phosphorylation of the retinoblastoma protein (pRb), ultimately promoting cell division. ERα can also promote cell cycle progression by binding to the C-terminal region of the p27 CDK inhibitor, preventing the nuclear accumulation of p27.Citation11 However, the mechanism through which ERα regulates cellular proliferation remains unclear for both normal and cancer cells. Some evidence suggests that inhibition of ERα by antiestrogens results in G0/G1 arrest;Citation12 however, we hypothesize a different mechanism by which both ERα and antiestrogens can modulate the cell cycle, where stabilization of ERα can delay cell cycle progression and ultimately lead to inhibition of cell proliferation.
ERα is an important regulator of growth and differentiation in normal breast tissues. Although most proliferating cells do not express ERα, the proportion of ERα-positive cells is increased in more highly proliferating structures.Citation13 Considerable evidence indicates that ERα also plays an important role in the development and progression of breast cancer. For example, clinical experience has established that ERα-positive breast tumors have a more favorable prognosis than breast tumors with little or no expression of ERα.Citation14-16 Therefore, the functional role of ERα expression in breast cancer etiology is paradoxical. The expression of ERα is associated with better prognosis, but ERα expression is low in normal mammary cells. In addition, triple-negative breast tumors have no ERα expression and are associated with a worse prognosis.Citation15 It is unclear whether the presence or absence of ERα provides a mechanism for cancer cell growth; therefore, ERα should not be classified as an oncogenic factor. The paradoxical nature of ERα also raises the question of how the presence or absence of ERα modulates the proliferation of cells in the context of the cell cycle.Citation11
The purpose of the present study was to elucidate the mechanism through which ERα influences cellular proliferation in human breast cancer cells. We examined the expression of ERα during the progression of cells through the various cell cycle phases in the presence or absence of its E2 ligand. We found a novel role for ERα in regulating the cell cycle and propose that ERα modulates the cell cycle by affecting the passage of cells through S and G2/M phases. These data also suggest a mechanism by which ligand-bound ERα leads to a faster cell cycle progression and unliganded ERα acts as a cell cycle inhibitor.
Results
ERα expression is cell cycle-regulated in synchronized MCF-7 cells
To determine the expression pattern of ERα during the cell cycle, we used MCF-7 cells synchronized in parallel under E2-negative and E2-positive conditions, harvested, and subjected to western blot and FACS analyses. When cells were released from lovastatin-mediated G1 arrest in the presence of E2, ERα protein levels increased at 24 hours, peaked at 28 hours, and eventually decreased after 40 hours (). We also examined levels of cyclin A and B proteins as markers of S and G2/M phases. Protein levels of cyclins A and B increased at 20 hours, peaked at 28 hours, and decreased after 40 hours, indicating a synchronous progression of the cell population after release from G1 arrest (). These data suggest that, in the presence of E2, ERα expression coincides with the expression of cyclins A and B, concomitant with the late S and G2/M phases of the cell cycle.
Figure 1. ERα is a cell cycle-regulated protein in synchronized MCF-7 cells. (A) MCF-7 cells were synchronized with lovastatin in the presence or absence of E2 and subjected to western blot analysis with indicated antibodies. (B) Quantification of ERα, cyclin A, and cyclin B protein levels relative to levels of actin in the presence or absence of E2.
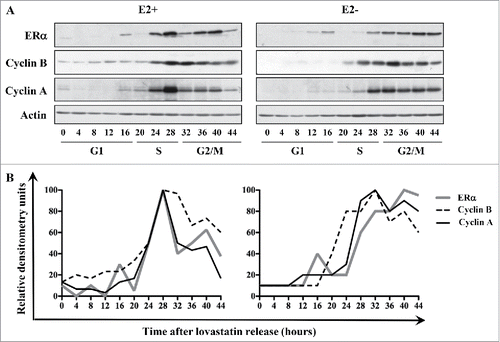
When MCF-7 cells were released from lovastatin-mediated G1 arrest in the absence of E2, ERα protein levels increased at 28 hours after release, as was the case in the presence of E2, however the levels of the protein never decreased and remained high until 44 hours (). Cyclins A and B showed a similar expression pattern: levels of cyclin A and cyclin B proteins increased at 28 hours and 24 hours, respectively, and levels did not notably decline over the observed time period (). Together, these results show that ERα is a cell cycle-regulated protein with peak expression during G2/M phase; however, the expression pattern is different depending on the liganded versus unliganded status of ERα.
Liganded ERα modulates cell cycle progression by decreasing the time MCF-7 cells spend in S and G2/M phases
Our findings that the presence or absence of E2 had a visible effect on the ERα expression pattern raised the possibility that the presence of ligand could modulate the length of the cell cycle in ERα-positive breast cancer cells. To address this question, we harvested lovastatin-synchronized MCF-7 cells cultured with or without E2 and subjected them to FACS analysis. The experiment was performed in triplicate, and data from all 3 replicates (I, II, and III) are depicted in . The results revealed a delay in the transition from S to G2/M when cells were cultured in the absence of E2. However, the length of G1 phase was very similar under E2-negative and E2-positive conditions in all 3 replicates. Quantitative analysis of the cell sorting data in replicate I revealed that 88% of MCF-7 cells were arrested in G1 phase and synchronously released into the cell cycle (). Synchronized MCF-7 cells exited G1 phase 17 hours after release under both E2-positive and E2-negative conditions; however, cells cultured under E2-negative conditions exhibited a delayed entry into the G1 phase of the next cell cycle compared with cells cultured under E2-positive conditions (). In replicate I, cells entered S phase approximately 4 hours earlier under E2-positive conditions than under E2-negative conditions, with entry times of 17 hours and 21 hours, respectively (). The decline of the S phase population was much sharper and entry into the subsequent S phase was observed earlier under E2-positive conditions than under E2-negative conditions (). Also, MCF-7 cells entered G2/M phase 7 hours earlier under E2-positive conditions than under E2-negative conditions (at 20 hours and 27 hours, respectively) and peaked 2 hours sooner (at 29 hours and 31 hours, respectively) (). Similar to the trend observed for S phase exit, G2/M exit was observed to be quicker under E2-positive conditions than under E2-negative conditions (). Also, entry into the subsequent G2/M phase was detectable under E2-positive conditions but not under E2-negative conditions because of the slower cell cycle progression ().
Figure 2. Liganded ERα hastens the passage through S and G2/M phases in MCF-7 cells. (A) MCF-7 cells were synchronized with lovastatin and released from G1 arrest with mevalonate in the presence of E2. DNA content was analyzed by PI staining, and cell cycle distribution was determined with CellQuest and ModFit software. (B) MCF-7 cells were synchronized with lovastatin in the presence or absence of E2, and DNA content was analyzed by PI staining. Three independent replicates are shown as I, II, and III. Hatched lines depict the start of the cell cycle phase.
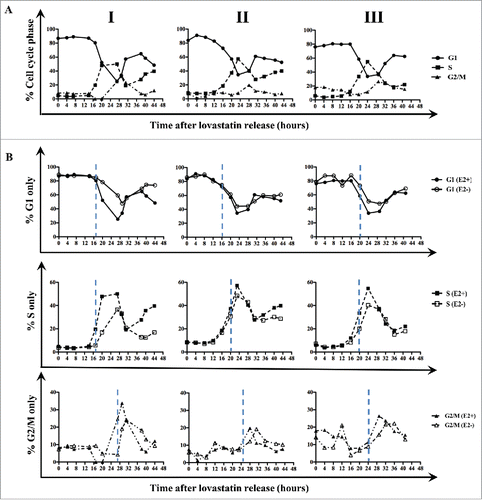
In the other 2 replicates (II and III), patterns of entry into and exit from S and G2/M phases were similar to those in replicate I, suggesting that the delay in entry into these phases could result in a longer cell cycle under E2-negative conditions. In fact, when the durations of the phases under liganded and unliganded conditions were determined from all 3 replicates, the quantification revealed that treatment of MCF-7 cells with E2 resulted in S and G2/M phases that were each shorter by 2.3 hours (). Consequently, the total cell cycle was longer under E2-negative conditions (36.5 hours) than under E2-positive conditions (32.0 hours); this difference was significant (P < 0.05). Overall, the results from all replicates raise the possibility that in MCF-7 cells, liganded ERα modulates cell cycle progression by shortening the S and G2/M phases of the cell cycle.
Table 1. Results of FACS analysis of (A) lovastatin-synchronized ERα-positive MCF-7 breast cancer cells in the presence or absence of E2 and of ERα-negative MDA-MB-231 breast cancer cells overexpressing ERα.; (B) lovastatin-synchronized MDA-MB-231 breast cancer cells overexpressing ERα; (C) lovastatin-synchronized MCF-7 cells treated with 10 μM E2 and 5 nM fulvestrant, 10 μM E2 alone, or 10 μM E2 and 5 μM tamoxifen.
Cell cycle regulation of ERα is not specific to the cell type or method of synchronization
We next asked whether the cell cycle expression pattern of ERα is specific to MCF-7 cells. To validate the cell cycle regulation of ERα, we used ZR75-1 cells, which exhibit similar phenotypic characteristics to MCF-7 cells: both are ERα -positive and PR-positive, have wild-type p53 and pRb, and have low invasive/metastatic potential. ZR75-1 cells were maintained in E2-free medium and subjected to lovastatin synchronization as described for MCF-7 cells. FACS analysis (Fig. S1A) revealed that lovastatin treatment effectively synchronized the cells by yielding a 95% arrest of ZR75-1 cells in G1, followed by synchronous progression through the cell cycle upon addition of mevalonate. Cells exited G1 at 15 hours after lovastatin release; the S phase fraction then increased, peaking at 20 hours. Cells exited S phase at 25 hours and then entered G2/M phase.
Western blotting revealed that ERα protein expression increased at 24 hours after release and remained high until after 40 hours (Fig. S1B). Cyclin B was also upregulated between 20 and 40 hours after release, a period coinciding with the S and G2/M phases observed in the FACS data (Fig. S1A and B). Western blotting and the corresponding densitometric results confirmed that ERα and cyclin B have similar patterns of expression during the cell cycle (Fig. S1B and C). These data suggest that endogenous ERα in ZR75-1 cells is subject to cell cycle regulation, as observed in MCF-7 cells, with peak expression during the S and G2/M phases.
To ensure that the cell cycle regulation of ERα was not specific to the method of synchronization, we also synchronized MCF-7 cells with aphidicolin which yields a G1/S arrest (Fig. S2A and B). Aphidicolin synchronization of MCF-7 cells revealed that the expression of ERα is cell cycle-regulated and that when E2 is present in the culture medium, a shortening of the cell cycle is observed (Fig. S2A and B). From this series of experiments, we can conclude that the cell cycle regulation of ERα is not a cell type-specific phenomenon, nor is it specific to the method of synchronization used.
ERα expression is not regulated at the transcriptional level
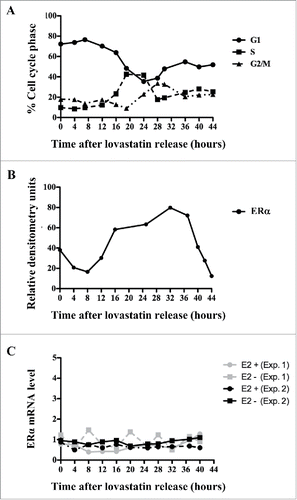
Although we had observed the cyclical expression of ERα protein by protein gel blotting ( and S1B), it remained unknown whether ERα mRNA was also regulated at the transcriptional level. To quantify the levels of ERα mRNA expressed throughout the cell cycle, we used MCF-7 cells synchronized with lovastatin and harvested at 4-hour intervals until 44 hours after release; RNA was extracted from cells at each time point. After reverse transcription, qRT-PCR was performed using sense and antisense oligos for ERα. FACS analysis revealed a similar pattern of synchronous cell cycle progression after lovastatin release to that observed here (). In contrast to the fluctuations of ERα protein levels (), qRT-PCR results showed that ERα mRNA levels did not change throughout the cell cycle (). Furthermore, ERα mRNA levels were unaffected by the presence or absence of E2 (). These data show that the cell cycle-dependent changes in ERα protein expression are not mediated via a transcriptionally regulated mechanism; therefore, we propose that the decrease in ERα expression during the G1 and early S phases is due to a post-transcriptional mechanism.
Figure 3. ERα is not regulated at the transcriptional level. MCF-7 cells were synchronized with lovastatin and subjected to: (A) PI staining to determine DNA content and cell cycle distribution; (B) quantification of ERα protein levels relative to actin expression under E2-positive conditions; and (C) qRT-PCR analysis to determine levels of ERα mRNA under E2-positive and E2-negative conditions.
Knockdown of ERα accelerates mitotic exit
The data presented thus far demonstrate that ligand-bound ERα modulates cell cycle progression by hastening the passage through the S and G2/M phases (). Therefore, we were interested in investigating whether knockdown of ERα in MCF-7 cells, under E2+ conditions, also affects cell cycle progression. To examine the efficacy of shRNA-ERα silencing, we used transiently transfected MCF-7 cells with the shRNA and harvested after 24, 36, and 48 hours. Western blotting indicated a decrease in ERα protein expression within 24 hours of transfection with shRNA-ERα (). Furthermore, ERα knockdown was maintained until 48 hours after transfection (). ERα protein levels remained constant in cells transfected with empty vector or scrambled control shRNA; however, upon transfection of MCF-7 cells with shRNA-ERα, ERα expression decreased (). The synchronization of ERα-knockdown MCF-7 cells was achieved by transfecting the cells with shRNA-ERα at the same time as the cells were plated for the synchronization, under E2+ conditions. Following release from lovastatin arrest, cells were harvested at time intervals coinciding with the S and G2/M phases (6–35 hours) and subjected to western blotting and FACS analyses. In synchronized parental MCF-7 cells, ERα protein levels increased at 24 hours, peaked at 29 hours, and decreased by 33 hours after release (). The MCF-7 cells transfected with shRNA-ERα did not show a detectable level of ERα for the duration of G2/M phase ().
Figure 4. Downregulation of ERα in MCF-7 cells hastens the passage through G2/M. MCF-7 cells were transfected with shRNA against ERα, scrambled control shRNA, or empty vector and subjected to: (A) immunoblotting with indicated antibodies; and (B) immunofluorescence staining to assess the efficacy of ERα knockdown. ERα is fluorescently labeled red, and the nuclei are fluorescently labeled blue with DAPI. MCF-7 cells were synchronized with lovastatin, in the presence of E2, transfected with shRNA-ERα, and subjected to: (C) western blotting with indicated antibodies; and (D) DNA content analysis by PI staining and subsequent FACS analysis to determine the impact of ERα silencing on cell cycle progression, particularly in G2/M phase.
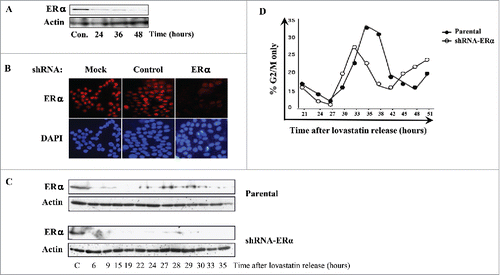
To detect potential effects of ERα knockdown on cell cycle progression through G2/M phase, we compared FACS analyses of the G2/M phases of parental and ERα-knockdown MCF-7 cells. The ERα-knockdown cells entered G2/M at 29 hours, peaked at 32 hours, and completely exited G2/M by 37 hours after release (). In contrast, parental MCF-7 cells entered G2/M at 29 hours, peaked at 34 hours, and slowly exited until 41 hours after release; therefore, mitotic exit was delayed by 4 hours for parental MCF-7 cells compared with ERα-knockdown cells (). In addition, FACS analysis showed that ERα-overexpressing MCF-7 cells exhibited prolonged S and G2/M phases compared with parental MCF-7 cells (Figs. S4A and B). Together, these results, which are consistent with those of , suggest that ERα plays a role in delaying mitotic exit.
ERα expression is cell cycle-regulated in synchronized MDA-MB-231 cells
Having shown that endogenous ERα is cell cycle-regulated in the ERα-positive cell lines MCF-7 and ZR75-1 ( and S1B), we next set out to determine whether exogenous ERα in ERα -negative breast cancer cells can also be subjected to cell cycle-dependent regulation. MDA-MB-231 cells were synchronized with lovastatin and achieved 80%–85% G1 arrest before synchronously progressing through the cell cycle after release. A pcDNA3.1 vector containing ESR1 was generated and transfected into the MDA-MB-231 cells 12 hours before release from arrest. FACS analysis reveals the synchronous progression of cells through the cell cycle (). Western blotting using whole-cell lysates from the synchronized cells demonstrated that cyclins A, B, and E were expressed during the cell cycle phase each is known to regulate (). Cyclin E was expressed between 12 and 24 hours after release, which corresponded with G1 phase as determined by FACS analysis (). Cyclin A was expressed at 20 hours after release, which corresponded to the onset of S phase, and cyclin B was upregulated starting 27 hours after release, corresponding to entry into G2/M phase ().
Figure 5. Exogenous ERα is cell cycle-regulated in ERα-negative breast cancer cells. MDA-MB-231 cells overexpressing exogenous ERα were cultured in complete alpha medium, synchronized with lovastatin and subjected to: (A) FACS analysis to determine the percentage of cells in each phase by PI staining and (B) western blotting for ERα and cyclins A, B, and E. (C) MDA-MB-231 cells expressing exogenous ERα were synchronized with lovastatin and released from G1 arrest with mevalonate in E2 free medium, but in the presence of E2. DNA content was analyzed by PI staining, and cell cycle distribution was determined by CellQuest and ModFit software. (D) MDA-MB-231 cells expressing exogenous ERα were synchronized with lovastatin in the presence or absence of E2, and DNA content was analyzed by PI staining. Three independent replicates are shown as I, II, and III. Hatched lines depict the start of the cell cycle phase.
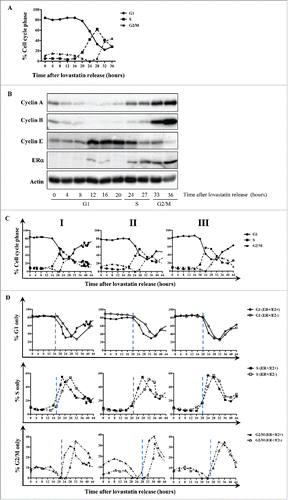
The percentage of cells in G1 declined sharply at 24 hours after release, when exogenous ERα was upregulated (). ERα protein levels remained high for the rest of the observed time period; therefore, the timing of ERα expression coincides with entry into the S and G2/M phases, as indicated by the expression of cyclins A and B (). In summary, ERα -negative MDA-MB-231 cells can also regulate the ectopic expression of ERα in a cell cycle-dependent fashion, similar to that of ERα -positive breast cancer cells, in which ERα expression is observed during S and G2/M phases.
ERα shortens S and G2/M phases of MDA-MB-231 cells
In MCF-7 cells, liganded ERα was shown to significantly decrease the time cycling cells spend in the S and G2/M phases compared with unliganded ERα, which delayed cell cycle progression ( and ). Therefore, it was critical to determine the effect of exogenous ERα on cell cycle progression in ERα-negative cells in the presence and absence of E2. Lovastatin-synchronized MDA-MB-231 cells were transiently transfected with ERα 12 hours before release from arrest; the E2-positive group received E2 at the same time. This experiment was performed in triplicate and analyzed by FACS ().
In all 3 replicates, lovastatin produced a high level of synchrony of cells through different phases of the cell cycle (). In replicate I, cells exited G1 at 17 hours under both conditions; however, the presence of E2 yielded a sharper transition out of G1 (). Entry into S phase was observed at 16 hours and 18 hours after release under E2-positive and E2-negative conditions, respectively (). In the presence of E2, a peak S phase fraction was observed at 22 hours and a subsequent exit began at 23 hours after release, whereas the absence of E2 delayed the S phase peak and exit until 28 hours and 32 hours, respectively (). Finally, MDA-MB-231 cells treated with E2 exhibited a premature G2/M phase compared with those cultured in the absence of E2. Specifically, E2-positive cells entered G2/M at 22 hours, peaked at 28 hours, and exited at 31 hours after release (), whereas the cells maintained under E2-negative conditions entered G2/M phase at 28 hours, peaked at 32 hours, and finally exited G2/M at 41 hours after release (). Results from replicates II and III agree with the findings from replicate I and show that in the presence of exogenous ERα, the S and G2/M phases are delayed when cells are maintained under E2-negative vs. E2-positive conditions (). Parental MDA-MB-231 cells were also synchronized in the absence of exogenous ERα, resulting in an inhibition of the cell cycle progression, similar to the effect of ERα overexpression on the cell cycle of MCF-7 cells (Fig. S5A and B). summarizes these results and shows the disparities between the cell cycles of parental MDA-MB-231 cells and ERα-expressing MDA-MB-231 cells maintained in the presence and absence of E2. In cells expressing exogenous, unliganded ERα, the duration of the S and G2/M phases was significantly shorter (by 9.1 hours, P < 0.05) compared with parental cells (). The cells maintained under liganded ERα conditions had significantly longer S and G2/M phases (by 2.0 hours, P < 0.05) compared with cells cultured under unliganded ERα conditions ().
Differential effects of tamoxifen and fulvestrant on cell cycle progression may be due to their unique interactions with ERα
Tamoxifen and fulvestrant are clinically used antiestrogens that exert their inhibitory effect on tumor proliferation by blocking the activity of ERα.Citation9 These drugs are distinct from one another in that fulvestrant promotes the degradation of ERα, whereas tamoxifen does not downregulate ERα protein. We used these 2 drugs as biochemical tools to investigate how either agent modulates the cell cycle expression pattern of ERα. We have already shown that downregulation of ERα has a stimulatory effect on the cell cycle and that the S and G2/M phases are prolonged when ERα is present without its E2 ligand (). Therefore, we hypothesized that fulvestrant would hasten cell cycle progression as a result of ERα degradation, whereas tamoxifen would not influence the cell cycle.
To test this hypothesis, we initially examined the expression of ERα in a dose-dependent manner by treating MCF-7 cells with increasing concentrations of these antiestrogens and subjecting them to western blot analysis to observe ERα expression. As expected, fulvestrant treatment of 2.5–80 nM resulted in the degradation of ERα in MCF-7 cells (). In contrast, tamoxifen treatment (2–8 μM) did not significantly affect the integrity of ERα expression, with only minimal downregulation of ERα at high doses (> 8 μM; ). Next, we examined the effects of tamoxifen and fulvestrant on the progression of cells through the cell cycle by synchronizing MCF-7 cells with lovastatin and treating them with either fulvestrant (5 nM) or tamoxifen (5 μM) in parallel experiments.
Figure 6. Effects of antiestrogens on cell cycle progression in MCF-7 cells. (A) MCF-7 cells were treated with increasing concentrations of fulvestrant (2.5–80 nM) and lysates were subjected to western blot analysis using the indicated antibodies. (B) MCF-7 cells were treated with increasing concentrations of tamoxifen (2–10 μM) and subjected to western blot analysis for the indicated antibodies. (C) MCF-7 cells were lovastatin-synchronized in the presence of E2 (10 μM), E2 and fulvestrant (5 nM), or E2 and tamoxifen (5 μM), and cells were stained with PI to determine DNA content. Cell cycle profiles were determined using CellQuest and ModFit software packages.
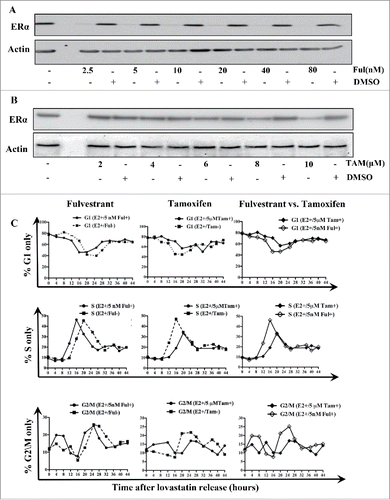
The results revealed that when MCF-7 cells were synchronized in the presence of E2, fulvestrant treatment resulted in faster progression through S and G2/M phases compared with untreated cells (). Specifically, in the presence of fulvestrant, MCF-7 cells exited G1 at 12 hours after release () and the subsequent S phase then began (). The peak S phase fraction was observed at 16 hours, and S phase exit began at 20 hours after release (). G2/M entry and exit occurred at 20 hours and 28 hours, respectively, and peak G2/M occurred at 25 hours after release (). Because fulvestrant promotes ERα degradation, these results are consistent with our previous conclusions that downregulation of ERα in ERα -positive breast cancer cells results in hastened progression through G2/M phase.
Unlike the results with fulvestrant, when synchronized cells were treated with tamoxifen, the progression through S and G2/M phases was much slower. Specifically, in lovastatin-synchronized MCF-7 cells treated with tamoxifen, the duration of S and G2/M was shorter by 3 hours compared with untreated cells, whereas in lovastatin-synchronized MCF-7 cells treated with fulvestrant, the duration of S and G2/M was shorter by 8 hours (; ). These data show that fulvestrant treatment of synchronized MCF-7 cells resulted in shorter S and G2/M phases compared with tamoxifen treatment ().
Discussion
We have found that ERα is cell cycle-regulated ( and S1) and also regulates the cell cycle as a function of E2. Specifically, we observed that the presence of E2 in the culture medium shortened the duration of the cell cycle (by 4.5 hours, P < 0.05) and that this discrepancy was due to decreased time spent in the S and G2/M phases (; ). Stimulation of breast cancer cells by estrogens has previously been shown to positively regulate proteins involved in the G1/S transition, such as c-Myc and cyclin D Citation12); therefore, it is noteworthy that in this study the G1 phase profiles were similar under E2-positive and E2-negative conditions ().
Our findings are consistent with those of earlier studies, which showed that expression of ERα in ERα -negative breast cancer cell lines results in growth inhibition under E2-positive conditions.Citation17-19 We observed that knockdown of endogenous ERα in ERα -positive cells accelerates mitotic exit () and that exogenous ERα expression in ERα -negative MDA-MB-231 cells shortens the S and G2/M phases compared with parental MDA-MB-231 cells, but to a lesser extent under liganded ERα conditions than under unliganded ERα conditions (; ).
We treated ERα-positive breast cancer cells with the antiestrogens tamoxifen and fulvestrant to modulate ERα expression in vitro, as is done in the clinical setting. Our data show that ERα degradation after fulvestrant treatment results in faster progression through S and G2/M phases compared with untreated cells, whereas tamoxifen treatment shortens the duration of S and G2/M phases by less than fulvestrant treatment does (). These observed differences in cell cycle progression can be understood by considering the differential effects of fulvestrant and tamoxifen on ERα protein stability. In MCF-7 cells transfected with shRNA-ERα, the S and G2/M phases are shorter because of the downregulation of ERα (), just as observed in the fulvestrant-treated MCF-7 cells (). In addition, MCF-7 cells treated with tamoxifen have a similar cell cycle profile to that of MDA-MB-231 cells with exogenously expressed ERα because tamoxifen treatment does not degrade the ERα protein (); therefore, the changes in transcription downstream of ERα play a role in stalling the S and G2/M phases upon tamoxifen treatment compared with fulvestrant treatment.
It would be reasonable to speculate that the combination of a drug that lowers estrogen levels, such as an aromatase inhibitor (AI), and an antiestrogen that does not decrease the levels of ERα, such as tamoxifen, would synergistically work to decrease proliferation of breast cancer cells by preventing ERα and E2 from stimulating cell cycle progression. Third generation AIs such as exemestane or the nonsteroidal anastrozole and letrozole have proved superior in both efficacy and tolerability compared to tamoxifen treatment in postmenopausal women with hormone receptor positive disease.Citation20 Fulvestrant has also shown to be effective in patients with ERα-positive advanced breast cancers with similar efficacy to tamoxifen.Citation10 Although the AIs and fulvestrant have distinct modes of action, the results from clinical trials comparing their effects on disease progression have concluded that there is no significant difference between these 2 classes of inhibitors. The studies presented in this report suggest that a more beneficial treatment strategy would be one that would combine a cell cycle inhibitor, such as a CDK2 inhibitor, followed by the combination of AI and tamxifen. Such sequential and combination treatment strategy would enrich the cells in a specific phase of the cell cycle (i.e. G1) to be most responsive to the combination of AI and tamxifen targeting E2 generation and ER activation.
Materials and methods
Cell cultures
ERα -positive (MCF-7 and ZR75-1) and ERα -negative (MDA-MB-231) cell lines were acquired from American Type Culture Collection. Forty vials of each cell line were frozen in liquid nitrogen before authentication by short tandem repeat analysis. Cultures were maintained in complete alpha medium supplemented with 1% of 1 mM HEPES (Sigma-Aldrich), 10% FBS (Atlanta Biologicals), 1% non-essential amino acids (Sigma-Aldrich), 1% of 200 mM glutamine (Sigma-Aldrich), 1% sodium pyruvate (Sigma-Aldrich), 0.1% of 1 mg/mL insulin (Sigma-Aldrich), 0.1% of 1 mg/mL hydrocortisone (Sigma-Aldrich), 0.05% of 25 μg/mL epidermal growth factor (Invitrogen), and 1% of 10 mg/mL ciprofloxacin (Bayer) in 37°C in a humidified incubator containing 6.5% CO2. The cells were passaged at a 1:10 ratio after 70%–80% confluency was achieved. To ensure the use of a consistent cell population, we cultured the contents of each vial for no more than 7 passages. New cultures were always derived from the 40 pre-authenticated vials.
To examine the effect of estrogen, we cultured MCF-7 cells in E2-free medium for no more than 3 weeks to completely clear the cells of estrogen. To prevent the slight estrogenic activity of the phenol red present in complete alpha medium, we used phenol red-free improved minimum essential medium (IMEM; Invitrogen) as described previously.Citation21 The IMEM was supplemented as described above, with 10% charcoal/dextran treated FBS substituted for the 10% FBS.
Synchronization of the cell cycle using lovastatin and aphidicolin
We used 2 different methods of synchronization to arrest the cells either in the early G1 phase, with lovastatin (as previously described; refs. Citation22,23), or at the G1/S phase boundary, with aphidicolin. When lovastatin maintains its closed lactone ring, it inhibits the 28S proteasome,Citation24 preventing the degradation of the p21 and p27 CDK inhibitors; therefore, CDK2 activity is inhibited and cells arrest in G1 with a high level of synchrony (85%; ref. Citation25). The resulting arrest is reversible upon the addition of mevalonate, leading to synchronous progression through the subsequent cell cycle phases. The lovastatin-mediated G1 arrest was achieved by treating cells with 10 μM lovastatin (LKT Laboratories) in E2-free medium for 36 hours. Cells were released from arrest by adding 1 mM mevalonate (Sigma-Aldrich), as previously described.Citation22 To assess the role of E2 in cell cycle progression, upon release from lovastatin arrest, we treated cells with or without 10 nM E2 simultaneously with 1 mM mevalonate. Cells were harvested at 4-hour intervals, starting at the time of release from arrest (referred to as 0 hours) until 44 hours after release.
Aphidicolin treatment was used to mediate a G1/S arrest, by selectively inhibiting DNA polymerase α.Citation26 This G1/S arrest was achieved by treating cells with 4 μg/mL aphidicolin (Sigma-Aldrich) for 24 hours. Cells were released from arrest by washing with E2-free medium. Cells were then harvested every 4 hours until 44 hours after release and prepared for western blot and fluorescence-activated cell sorting (FACS) analyses. E2 (10 nM) was added to the E2-positive arm at the time of release from arrest. Experiments comparing E2-positive and E2-negative conditions were always run in parallel.
Western blotting and antibodies
Whole-cell lysates were prepared for protein gel blot analysis as previously described.Citation27 The monoclonal antibodies used were against ERα (Novocastra Laboratories), cyclin A, cyclin B, cyclin E (all from Santa Cruz Biotechnology), and actin (C4, Chemicon), and antibodies were prepared in blocking buffer at 1 μg/mL. Incubation for 1 hour with horseradish peroxidase-conjugated secondary antibody (Pierce Biotechnology) at 1:5000 in blocking buffer was followed by detection using a chemiluminescence reagent (NEN Life Science Products) as directed by the manufacturer. Densitometric analyses were performed, relating the band intensities of the probed protein to those of actin.
Cell cycle analysis
For DNA content analysis, 1 × 106 cells were harvested and washed with cold PBS. After centrifugation at 1400 rpm for 4 minutes at RT to pellet the cells, cells were fixed overnight with 60% ethanol at 4°C. Cells were then washed twice with PBS and stained with a solution containing 10 μg/mL propidium iodide (PI), 20 μg/mL RNase A, 0.5% Tween 20, and 0.5% bovine serum albumin (BSA) in PBS (all reagents from Sigma-Aldrich). Cells were filtered 24 hours after the staining and subjected to analysis using a FACSCalibur flow cytometer (Becton Dickinson). The percentage of cells in each phase of the cell cycle was analyzed using CellQuest software (Becton Dickinson) and was statistically analyzed using ModFit software (Verity Software House). A diploid model was selected, and the position and range of the marker placement were assessed. The “fit” option was also selected to calculate the relative cell cycle distribution (G1, S, G2/M) of the acquired cells. The duration of each phase was defined as the time elapsed from when the percentage of cells started to increase in a given phase (ascending line) until the percentage of cells started to decrease in the same phase (descending line). The duration of each cell cycle phase is expressed as the mean ± standard deviation from 3 independent replicate experiments. The statistical significance of the difference in the duration of phases in E2-positive and E2-negative conditions was measured using a 2-sided Student's t test and a significance level of 5% (P < 0.05).
RNA extraction and quantitative real-time PCR
Total RNA was extracted from synchronized MCF-7 cells using guanidinium isothiocyanate homogenization followed by cesium chloride centrifugation, as previously described.Citation28 Quantitative real-time PCR (qRT-PCR) was carried out on an ABI 7500 system (Applied Biosystems). The relative gene expression was determined using the comparative CT method and normalized to the housekeeping gene cyclophilin. The ERα-specific primer used was 5′-TGG GCT TAC TGA CCA ACC TG-3′; 5′-CCT GAT CAT GGA GGG TCA AA-3′. Each qRT-PCR assay was performed in triplicate, and the result was confirmed in 2 independent experiments.
Knockdown of ERα with shRNA
ERα short hairpin RNA (shRNA) was inserted into the pSilencer 2.0-U6 expression vector (Ambion) according to protocols provided by the manufacturer. The DNA oligos for ERα shRNA vector construction were synthesized by Sigma-Genosys. The sense oligo sequence was 5′-GAT CCC GCG CTC TAA GAA GAA CAG CCT TCA AGA GAG GCT GTT CTT CTT AGA GCG TTT TTT GGA A-3′, and the antisense oligo sequence was 5′-AGC TTT TCC AAA AAA CGC TCT AAG AAG AAC AGC CTC TCT TGA AGG CTG TTC TTC TTA GAG CGC GG-3′. For ERα knockdown, MCF-7 cells were cultured on a 100-mm culture dish to 70% confluency in complete alpha medium (i.e., in the presence of E2) with 10% FBS. ERα shRNA or negative control vector (Ambion) at 10 μg/dish was premixed with 15 μL of FuGene6 (Roche) and added to each dish. Cells were harvested at the indicated times after transfection and subjected to western blot and immunocytochemical analyses with an antibody against ERα.
Immunocytochemical staining
Cells (5 × 104) were plated on sterilized cover slips that had been placed inside each well of a 6-well plate. At the time of harvest, cells on the coverslips were fixed with 4% paraformaldehyde for 5 minutes at RT, washed with PBS 3 times for 5 minutes each, and blocked with 3% BSA containing 0.1% NP-40 in PBS for 1 hour at RT. Primary antibodies were added at 1:200 in blocking buffer and were incubated on the slides overnight at 4°C. The next day, the slides were washed with PBS 3 times for 10 minutes each and incubated with fluorescently labeled secondary antibodies for 3 hours at RT. Slides were washed again with PBS 3 times for 10 minutes each and then covered with 4′,6-diamidino-2-phenylindole (DAPI) containing antifade solution (Vector Laboratories). Slides were analyzed using an Olympus DSU spinning disc confocal fluorescence microscope.
Overexpression of ERα
To evaluate the effects of overexpression of ERα, we arrested MCF-7 and ERα-negative MDA-MB-231 cells in the early G1 phase by treatment with lovastatin and released them from arrest by treatment with mevalonate, as described above. Cells were transfected with human pSG5-ERα or control vector at the time of mevalonate addition. Cells were harvested at the indicated time points for western blot and FACS analyses.
Treatment of cells with fulvestrant and tamoxifen
Asynchronous populations of MCF-7 cells were plated at 1 × 106 cells/plate and treated with increasing concentrations of fulvestrant (2.5–80 nM) or tamoxifen (2–10 μM) for 24 hours. Cells were harvested and subjected to western blot analysis with an antibody against ERα. To investigate the effects of these antiestrogens on cell cycle transition, we also subjected G1-arrested populations of cells to treatment with tamoxifen (5 μM) or fulvestrant (5 nM) for 0–44 hours after lovastatin release. Cells were harvested at 4-hour intervals and subjected to FACS analysis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1166327_Supplemental_Material.zip
Download Zip (1.8 MB)Acknowledgments
We thank Dr. Rozita Bagheri-Yarmand for helpful discussion during the course of this work, Dr Joseph (Tony) Caruso for help with the critical reading of the manuscript, Mr. Tuyen Bui for his technical expertise and Ms. Karen Muller in the Department of Scientific Publications for editorial review of this manuscript.
Funding
This work was supported in part by the DOD Predoctoral Traineeship award (W81XWH-06-1-0339) to SJM, the National Cancer Institute (CA87548 and CA1522218) to KK, and the MD Anderson Cancer Center Core Grant (CA016672).
Related Research Data
References
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev 2001; 81:1535-65; PMID:11581496.
- MacGregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol Reviews 1998; 50:151-96; PMID:9647865.
- Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, Santen RJ. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol 2002; 16:116-27; PMID:11773443.
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000; 407:538-41; PMID:11029009.; http://dx.doi.org/10.1038/35035131
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006; 295:2492-502; PMID:16757721.; http://dx.doi.org/10.1001/jama.295.21.2492
- Calza S, Hall P, Auer G, Bjohle J, Klaar S, Kronenwett U, Liu ET, Miller L, Ploner A, Smeds J, et al. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res 2006; 8:R34; PMID:16846532.
- Devin-Leclerc J, Meng X, Delahaye F, Leclerc P, Baulieu EE, Catelli MG. Interaction and dissociation by ligands of estrogen receptor and Hsp90: the antiestrogen RU 58668 induces a protein synthesis-dependent clustering of the receptor in the cytoplasm. Mol Endocrinol 1998; 12:842-54; PMID:9626660.; http://dx.doi.org/10.1210/mend.12.6.0121
- Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A 1992; 89:4037-41; PMID:1570330.; http://dx.doi.org/10.1073/pnas.89.9.4037
- Wakeling AE. The future of new pure antiestrogens in clinical breast cancer. Breast Cancer Res Treat 1993;25:1-9; PMID:8518404.; http://dx.doi.org/10.1007/BF00662395
- Howell A, Robertson JF, Abram P, Lichinitser MR, Elledge R, Bajetta E, Watanabe T, Morris C, Webster A, Dimery I, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol 2004; 22:1605-13; PMID:15117982.
- JavanMoghadam S, Hands AM, Keyomarsi K. Breaking the cycle: an insight into the role of ERα in eukaryotic cell cycles. J Carcinog 2011; 10:25; In Press; PMID:22190867.
- Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer 2003; 10:179-86; PMID:12790780.; http://dx.doi.org/10.1677/erc.0.0100179
- Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat 1999; 53:217-27; PMID:10369068.; http://dx.doi.org/10.1023/A:1006186719322
- Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A 2003; 100:10393-8; PMID:12917485.; http://dx.doi.org/10.1073/pnas.1732912100
- Podo F, Buydens LM, Degani H, Hilhorst R, Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J, Monleon D, et al. Triple-negative breast cancer: present challenges and new perspectives. Mol Oncol 2010;4:209-29; PMID:20537966.; http://dx.doi.org/10.1016/j.molonc.2010.04.006
- Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer 2004; 40:2667-75; PMID:15571950.; http://dx.doi.org/10.1016/j.ejca.2004.08.021
- Jiang SY, Jordan VC. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. J Natl Cancer Inst 1992;84:580-91; PMID:1556769.; http://dx.doi.org/10.1093/jnci/84.8.580
- Platet N, Cunat S, Chalbos D, Rochefort H, Garcia M. Unliganded and liganded estrogen receptors protect against cancer invasion via different mechanisms. Mol Endocrinol 2000; 14:999-1009; PMID:10894150.; http://dx.doi.org/10.1210/mend.14.7.0492
- Garcia M, Derocq D, Freiss G, Rochefort H. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. Proc Natl Acad Sci U S A 1992; 89:11538-42; PMID:1454845.; http://dx.doi.org/10.1073/pnas.89.23.11538
- Wong ZW, Ellis MJ. First-line endocrine treatment of breast cancer: aromatase inhibitor or antioestrogen? Br J Cancer 2004; 90:20-5; PMID:14710200.
- Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, Tucker SL, Chang S, Keyomarsi K. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res 2004; 64:3198-208; PMID:15126360.; http://dx.doi.org/10.1158/0008-5472.CAN-03-3672
- Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res 1991; 51:3602-9; PMID:1711413.
- Javanmoghadam-Kamrani S, Keyomarsi K. Synchronization of the cell cycle using lovastatin. Cell Cycle 2008; 7:2434-40; PMID:18677105.; http://dx.doi.org/10.4161/cc.6364
- Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci U S A 1999; 96:7797-802; PMID:10393901.; http://dx.doi.org/10.1073/pnas.96.14.7797
- Rao S, Lowe M, Herliczek TW, Keyomarsi K. Lovastatin mediated G1 arrest in normal and tumor breast cells is through inhibition of CDK2 activity and redistribution of p21 and p27, independent of p53. Oncogene 1998; 17:2393-402; PMID:9811471.; http://dx.doi.org/10.1038/sj.onc.1202322
- Pedrali-Noy G, Spadari S, Miller-Faures A, Miller AO, Kruppa J, Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Res 1980; 8:377-87; PMID:6775308.; http://dx.doi.org/10.1093/nar/8.2.377
- Keyomarsi K, Conte D, Jr., Toyofuku W, Fox MP. Deregulation of cyclin E in breast cancer. Oncogene 1995; 11:941-50; PMID:7675453.
- Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A 1993; 90:1112-6; PMID:8430082.; http://dx.doi.org/10.1073/pnas.90.3.1112