
ABSTRACT
The p300-mediated acetylation of enzymes involved in DNA repair and replication has been previously shown to stimulate or inhibit their activities in reconstituted systems. To explore the role of acetylation on DNA repair in cells we constructed plasmid substrates carrying inactivating damages in the EGFP reporter gene, which should be repaired in cells through DNA mismatch repair (MMR) or base excision repair (BER) mechanisms. We analyzed efficiency of repair within these plasmid substrates in cells exposed to deacetylase and acetyltransferase inhibitors, and also in cells deficient in p300 acetyltransferase. Our results indicate that protein acetylation improves DNA mismatch repair in MMR-proficient HeLa cells and also in MMR-deficient HCT116 cells. Moreover, results suggest that stimulated repair of mismatches in MMR-deficient HCT116 cells is done though a strand-displacement synthesis mechanism described previously for Okazaki fragments maturation and also for the EXOI-independent pathway of MMR. Loss of p300 reduced repair of mismatches in MMR-deficient cells, but did not have evident effects on BER mechanisms, including the long patch BER pathway. Hypoacetylation of the cells in the presence of acetyltransferase inhibitor, garcinol generally reduced efficiency of BER of 8-oxoG damage, indicating that some steps in the pathway are stimulated by acetylation.
KEYWORDS:
Introduction
Acetylation of lysine (K) residues is an important modification in defining protein function through altering its structure, activity, stability or ability to interact with other biomolecules. Addition of an acetyl group to lysine residues is reversible and was initially described as an epigenetic modification of histones regulating chromatin structure and transcription. Currently, acetylation is recognized as a major post-translational modification of non-histone proteins modulating different biological processes such as gene expression, cell proliferation, cell survival and apoptosis.Citation1-3 The importance of this protein modification is emphasized by the fact that some cancers are associated with deregulated acetylation of lysine residues in histones, transcription factors or in critical regulatory proteins, such as tumor suppressor p53 or proto-oncogene Ras.Citation4-7
Whereas the influence of acetylation on the structure of chromatin has been well defined, recent studies indicate that acetylation also regulates activity of DNA repair proteins. The link between acetylation and DNA damage repair was first detected in mammalian cells with induced hyperacetylation after exposure to a deacetylase inhibitor.Citation8-10 Since the treatment has an effect on chromatin structure, it was debated whether the observed increase in excision repair of UV-damaged DNA resulted from better accessibility of damage sites to repair enzymes in relaxed chromatin or other factors. It is now evident that acetylation of both histone and non-histone proteins regulates pathways of DNA repair. For example, certain patterns of acetylated K residues in histones are essential in repair of broken DNA, suggesting that a particular pattern of modifications acts as a signal to DNA repair factors.Citation11-13 In the case of non-histone proteins, results indicate that acetylation of specific K residues regulates activity of enzymes involved in DNA repair. For example, acetylation of DNA polymerase β (Pol β) impaired its dRP-lyase activity essential in short-patch base excision repair (SP-BER) when tested using a reconstituted assay.Citation14 Moreover, acetylation of the Werner DNA helicase (WRN) stimulated Pol β-mediated strand displacement DNA synthesis in vitro, suggesting up-regulation of the long patch (LP) BER pathway in cells upon acetylation.Citation15 A positive effect of acetylation on strand displacement synthesis was also observed in reconstituted systems simulating Okazaki fragments synthesis.Citation16-18 The effect of acetylation on proteins involved in DNA mismatch repair (MMR) was investigated in less detail. Here, acetylation of K residues at the C termini of MSH2, an especial component of the complex recognizing mismatches in DNA, had a positive effect on repair.Citation19 Interestingly, deacetylation of N termini in MSH2 also had a positive effect on MMR in reconstituted systems. Citation20
In this study, our goal was to determine the effect of acetylation on DNA repair mechanisms in cell-based systems using plasmid constructs bearing damage sites in the EGFP gene and monitoring its repair. We focused on analyzing two DNA repair mechanisms, MMR and BER. Our results demonstrate for the first time that acetylation enhances repair of mismatches in HeLa cells and in HCT116 cells, deficient in MMR. We also demonstrate that loss of p300 reduces efficiency of mismatch repair in cells deficient in MMR. The loss of p300 did not affect repair of 8-oxoG, although hypoacetylation reduced repair of this damage. Our results also suggest that repair of mismatches in MMR-deficient cells is performed through an acetylation-regulated EXOI-independent pathway involving enhancement of a strand-displacement synthesis mechanism previously described in reconstitution of lagging strand DNA replication.
Results
Principle of the DNA repair assay in vivo
To determine the effects of cellular protein acetylation capacity on DNA repair in mammalian cells we utilized plasmid DNA constructs containing an enhanced green fluorescent protein (EGFP) gene harboring alterations or damage diagnostic for characteristics of DNA repair. The cells were transfected with the constructs and EGFP expression was monitored by flow cytometric analysis. We generated two series of plasmids modified in the transcribed strand of the EGFP gene: 1) Constructs with nicks and a mismatch creating a stop codon positioned at various locations from the nick (). The presence of nicks ensures discrimination of DNA strands, so that repair of the mismatch is specific for the transcribed strand. 2) Constructs with a damaged base (8-oxoG; 8-oxo-7,8-dihydroguanine) and with a mismatch creating stop codon positioned on the 3′ side of the lesion (). Three human cell lines were used for transfection, HeLa, HCT116, deficient in DNA mismatch repair, and HCT116 derivative cells containing a knockout of acetyltransferase p300. The observed effects allowed us to establish whether acetylation influences DNA repair mechanisms in cells.
Figure 1. Illustration of plasmid substrates used to analyze mechanisms of DNA repair in a cell-based system. Plasmid constructs were made with nicks (marked N, position +1 for 5′N) (A), or with 8-oxoG lesion (marked X, position +1) (B), and with or without a mismatch creating a stop codon (star), at varying distances from the lesion or the nick. NT, non-transcribed strand. T, transcribed strand. (C) Outline of the strategy used for construction of the plasmids. The procedure was adopted from Kitsera et al. Citation30 Two restriction sites recognized by the nicking enzyme Nb.BbvCI were inserted into the ORF of EGFP to efficiently remove the native single-stranded DNA fragment (i). The single-stranded DNA fragments of the transcribed template excised by the nicking endonuclease were sequentially exchanged for the matching synthetic oligonucleotides containing the modification, 8-oxoG and/or single base alteration creating a stop codon in N-terminal EGFP (ii). The new oligonucleotide annealed to a gapped plasmid was ligated or left unsealed (iii). (D) Generation of gapped plasmids by Nb.BbvCI (left panel, lane 3). Nicking of plasmid results in formation of an open circular (oc) form, which migrates in the ethidium bromide agarose gel slower than covalently closed (ccc) untreated plasmid (lane 1), and linear form (lane 2) of plasmid from treatment with HindIII. Presence of 180-fold molar excess of the competitor oligonucleotides efficiently replaces native single-stranded DNA fragment (right panel). Exchange of the 5′-phosphorylated native DNA fragments with unphosphorylated synthetic oligonucleotides is detected by the inhibition of ligation in the absence of PNK. Reactions performed in the presence of PNK prior ligations result in covalently closed (ccc) plasmid.
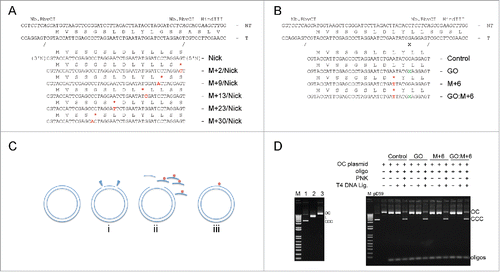
All plasmid constructs were produced by annealing of a synthetic oligonucleotide containing the desired modification to a circular plasmid DNA with a pre-created complementary single-stranded gap (). To generate the gap, we first constructed plasmid substrates containing two sites recognized by a sequence-specific nicking enzyme Nb.BbvCI. The cleavage sites were placed so that only a selected region of the transcribed strand of the EGFP gene was excised. The original single-stranded DNA between the two Nb.BbvCI sites was melted out and replaced by the synthetic oligonucleotide. For plasmid constructs with an incorporated damaged base, the nicks were ligated and then the DNA was subjected to CsCl gradient purification. For experiments using nicked plasmids, the nicks were left unsealed. Successful replacement of the native sequence with the synthetic oligonucleotide was determined by monitoring inhibition of ligation of the dephosphorylated strands in control reactions in the absence of T4 DNA polynucleotide kinase ().
DNA repair analyses with plasmids containing nicks or nicks+mismatch
Nicked plasmids with a mismatch () are likely substrates for various DNA repair mechanisms. In cells, the presence of a nick allows DNA mismatch repair (MMR) mechanisms to fix errors introduced during DNA replication, recombination or even other pathways of repair. During synthesis of the lagging strand in DNA replication, nicks between Okazaki fragments direct the mismatch-proofreading systems of MMR specifically to correct errors introduced into the newly-synthesized daughter strand. In cells, once a DNA mismatch is identified the repair is done with or without involvement of EXO1. The proposed alternative EXO1-independent MMR pathway with strand displacement synthesis mechanism was analyzed in detail using reconstituted systems (). Citation21-24
Figure 2. Diagram of DNA mismatch repair (MMR) and base excision repair (BER) pathways.
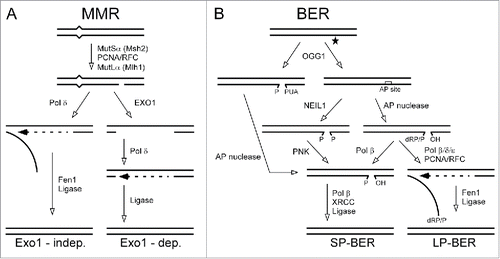
The colon carcinoma cell line HCT116 used here is deficient in MMR. Citation25 When the modified plasmids are transfected into these cells, we consider that nicked DNA constructs with a mismatch become primary substrates for an EXO1-independent path proposed to involve DNA replication enzymes (). Citation26 Thus, in this setting, repair of the mismatch near a nick is likely to result from Pol δ - directed synthesis involving strand displacement with support of PCNA. Placement of the mismatch at various locations from the nick would largely report on effects of protein acetylation on mechanisms linked to this synthesis-driven displacement.
DNA repair analysis with damaged base-containing plasmids
Plasmid constructs containing a damaged base () will attract cellular factors involved in DNA base excision repair (BER). The 8-oxoG lesion is a major product of DNA oxidation and one of the most frequent mutagenic damaged bases. BER is accomplished through two alternative pathways (). In short patch BER the damaged base is excised and the resulting one-nucleotide gap is filled and ligated. Long patch BER involves significant strand displacement synthesis before ligation.
To monitor LP-BER we used a system developed by Sattler et al.Citation27 Here, a mismatch creating a stop codon was placed on the 3′ side of the lesion (). If constructs are transfected into MMR-deficient HCT116 cells, repair of this mismatch depends on strand displacement synthesis utilized in LP-BER. Since the plasmids are covalently closed and MMR incision in HCT116 is impaired, little MMR should occur. Repair at the lesion by the SP-BER pathway will not extend to the mismatch. Repair of the mismatch should only reflect that the damaged base was repaired through the LP-BER pathway, with the replaced patch extended to the mismatch. As a reference sample in the LP-BER analysis, we used a plasmid construct with a mismatch-creating stop codon, but without the lesion.
Directly monitoring SP-BER is more complicated. While the plasmid with the damaged base and nearby mismatch-creating stop codon will exclusively monitor LP-BER, the plasmid with only a damaged base will be repaired by either SP- or LP-BER. In addition, the presence of 8-oxoG in the transcribed strand does not strongly inhibit transcription. Thus, the unrepaired base will still result in EGFP expression.Citation28,29 However, inefficient repair of 8-oxoG leading to accumulation of single-strand break intermediates will affect EGFP transcription, as previously demonstrated by Kitsera et al.Citation30 Thus, plasmid with a single damaged base 8-oxoG, to some extent, will monitor the effect of acetylation on both BER pathways.
Efficiency of MMR increases in cells exposed to a histone deacetylase inhibitor
To determine how protein acetylation influences DNA repair in cells, we first examined repair of our plasmid constructs in the presence of sodium butyrate, a histone deacetylase inhibitor (HDACi) that augments protein acetylation. Sodium butyrate is one of the most widely studied HDACi compounds, and was previously shown to increase efficiency in excision repair of UV-damaged DNA.Citation8,10,31 Since butyrate inhibits deacetylation of histones, it has an effect on chromatin structure. If so, increased efficiency of DNA repair could be a result of more relaxed chromatin allowing DNA repair enzymes to have better access to damaged sites. Nucleosomes are also formed on plasmids, but association of histones was found to be anomalous and plasmid DNA did not form closely packed nucleosomes, as observed in cellular chromatin.Citation32 Thus, we considered that butyrate-related effects on DNA repair within plasmid constructs would likely not result primarily from changes in associations of the plasmid DNA with histones, and instead more directly report on the influence of acetylation on repair protein functions.
To determine how hyperacetylation of proteins in HeLa cells affects DNA repair of our plasmid constructs, we first treated the cells with sodium butyrate at two different concentrations, and 6 hours later performed transfection with undamaged plasmid (Control), nick-containing plasmid (Nick) and a plasmid with a mismatch placed 13nt away on the 3′ side of the nick (M+13/Nick) (). The level of EGFP expression in the absence of butyrate was reduced for the construct with nicks and even more for the construct with nicks and a mismatch-creating a stop codon, compared to undamaged plasmid (). When cells were exposed to HDACi, the relative EGFP expression from plasmids with lesions increased up to 1.45 fold () as compared to untreated cells and undamaged Control.
Figure 3. Effects of sodium butyrate on DNA repair of nicks- and nicks+mismatch-containing plasmids in HeLa (A) and HCT116 cells (B). Cells were first exposed to different concentrations of sodium butyrate and 6 h later were transfected with undamaged plasmid (Control), nicks-containing plasmid (Nick) and the plasmid with a mismatch placed 13nt away on the 3′ side of the nick (M+13/Nick). 24 h after transfection the positive EGFP population was analyzed by flow cytometry. The presented data are the mean of 3 independent experiments; error bars indicate SD. The star (*) indicates P < 0.05 using a Student's t-test and represent a statistically significant difference. P values were compared with the culture without inhibitor.
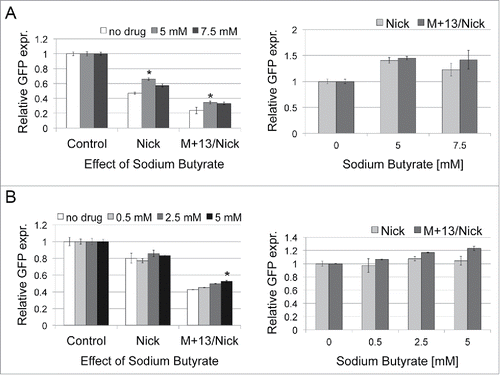
Relative increases in the EGFP positive population in the presence of HDACi were also observed in HCT116 cells with deficient MMR, but only for the plasmid construct M+13/Nick. The effects were smaller than with HeLa cells, up to 1.2-fold at 5 mM sodium butyrate (). HCT116 has a defective MLH1 gene expressing a component of MutLα, an endonuclease directed by PCNA to perform incision near a mismatch.Citation33 In our system, nicks are already present in the plasmid construct. MLH1 is also involved in recruitment of EXO1.Citation34 With the defective MLH1 gene, formation of the MMR complex is likely impaired in HCT116, and the M+13/Nick construct might become a substrate for repair through the EXO1-independent MMR mechanism, proposed to use enzymes involved in synthesis of Okazaki fragments. Thus, we hypothesize that this setting reflects strand displacement synthesis, with PCNA loaded at a nick and forming a complex with Pol δ. Here, the mismatch-creating stop codon is removed through formation of a flap subsequently cleaved as described in Okazaki fragment maturation and EXO1-independent MMR.
Overall, these results indicate that repairing of nicks in the transcribed strand of the EGFP gene is more efficient in cells with an elevated level of acetylation. This observation suggests that enzymatic activity or interactions of DNA ligase, PCNA, or other components involved in DNA sealing are enhanced by acetylation. Interestingly, for the construct with nicks and a mismatch, the relative increases in EGFP expression are even greater than for the construct with nicks only, suggesting that acetylation has a positive effect on DNA repair mechanisms linked to removal of the mismatch. These differences in DNA repair also indicate that acetylation-related effects are directly or indirectly linked to mechanisms of DNA repair of these lesions, and not to changes in structure and accessibility of the reporting plasmid.
Efficiency of MMR is reduced in cells exposed to an acetyltransferase inhibitor
We next investigated whether exposing cells to an inhibitor of lysine acetyltransferase would affect DNA repair of our plasmid constructs. Garcinol is a polyisoprenylated benzophenone derivative from rinds of Garcinia indica fruit. It inhibits the activities of histone acetyltransferases (HATs), including CBP/p300 and PCAF.Citation35 The effect of garcinol on DNA repair was previously analyzed in HeLa cells, where it was demonstrated that it inhibited non-homologous end joining (NHEJ), a major pathway used to repair DNA double-strand breaks (DSBs).Citation36 Since garcinol inhibits acetylation of histones, it also has an effect on structure of chromatin. It was proposed that the inhibitory effect of garcinol on NHEJ is linked to chromatin compacting resulting from blocked acetylation, impeding chromatin accessibility at sites of DNA breaks.
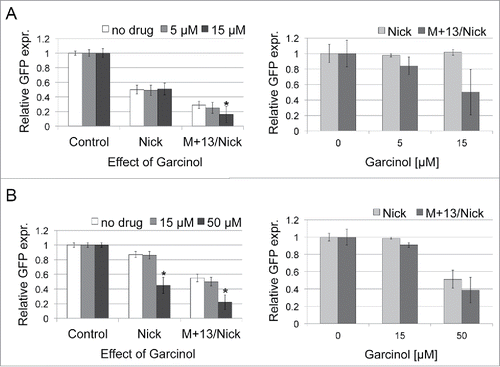
To determine how garcinol affects DNA repair of our plasmid constructs, we treated HeLa and HCT116 cells with the inhibitor at two different concentrations, and 2 hours later performed transfection with undamaged plasmid (Control), plasmid with nicks (Nick) and plasmid with a mismatch placed 13nt away on the 3′ side of a nick (M+13/Nick) (). Garcinol reduced the proportion of EGFP-positive cells in a dose-dependent manner, although the pattern of reductions was different in comparing the two cell lines. HeLa cells showed lower tolerance to high concentration of inhibitor (50 μM), previously reported to induce apoptosis (data not shown).Citation35 Garcinol treatment at 15 μM showed no effect on repair of the nicked plasmid in both cell lines, whereas the repair of the nicks+mismatch-containing plasmid was reduced about 50% in HeLa, and 10% in HCT116 cells. The drug had no influence on DNA repair of the nicks-containing plasmid, but its inhibitory effect on a repair of nicks+mismatch plasmid indicates that observed effects are directly or indirectly linked to DNA repair of these lesions, and are not related to association of nucleosomes with plasmid DNA. Exposure of HCT116 to 50 μM of garcinol resulted in inhibition of EGFP expression with both plasmid constructs, up to 50–60% (). Overall results indicate that protein acetylation improves repair of nicks and mismatches, even when MMR is defective.
Figure 4. Effects of garcinol on DNA repair of nicks- and nicks+mismatch-containing plasmids in HeLa (A) and HCT116 cells (B). Cells were treated as described in . The presented data are the mean of 4 independent experiments; error bars indicate SD. The star (*) indicates P < 0.05 using a Student's t-test. P values were compared with the culture without inhibitor.
Efficiency of BER is not significantly altered in cells exposed to deacetylase and acetyltransferase inhibitors
We next assessed the effects of sodium butyrate and garcinol on BER mechanisms. Here, M+6 is a DNA construct with a mismatch-creating stop codon in the transcribed strand of EGFP in a covalently closed plasmid. A single nucleotide mismatch was introduced at the 3′ side of an undamaged G residue, located 6 nucleotides away (). Since MMR mechanisms are impaired in HCT116 cells, the efficiency of repair for this mismatch is reduced to about 20% that of unaltered gene. Moreover, in plasmid construct GO:M+6 (), the G residue was replaced with a damaged base, 8-oxoG, allowing for recruitment of BER proteins that would repair both the damaged base and incidentally replace a long enough patch to improve rescue of EGFP expression (). The removal of the mismatch occurs here through the LP-BER mechanism involving strand displacement synthesis by Pol β/δ/ε, initiated at the damaged base after its excision. Thus, the rescue of mismatch repair in the GO:M+6 construct reflects LP-BER. The plasmid construct GO contains only a damaged base, and its repair is not necessary for EGFP expression, since unrepaired 8-oxoG does not cause a transcriptional arrest.Citation28,29
Figure 5. Effects of sodium butyrate (A) and garcinol (B) on BER in HCT116 cells. Cells were treated as described in . Two plasmid constructs, M+6 and GO:M+6 were used to monitor LP-BER. The general effects of inhibitors on BER are assessed with the GO construct. The presented data are the mean of 3 independent experiments; error bars indicate SD. The star (*) indicates P < 0.05 using a Student's t-test. P values were compared with the culture without inhibitor.
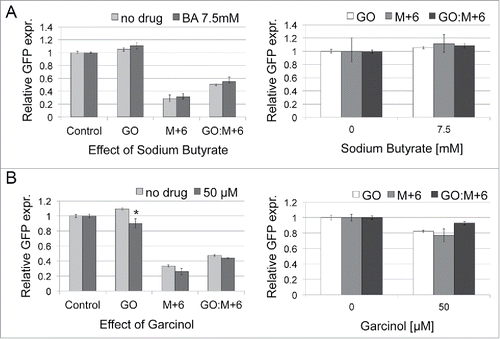
As shown in , neither sodium butyrate nor garcinol had an influence on the rescue of mismatch repair through the LP-BER mechanism. Interestingly, an inhibitory effect of garcinol was observed for the GO plasmid construct, although repair of 8-oxoG should not have influence on EGFP expression. Previous reports indicated that although unrepaired 8-oxoG does not prevent transcription, a single-strand break generated after the 8-oxoG excision inhibits transcriptionCitation30. Thus, the inhibitory effect of garcinol might result from accumulation of intermediates that are not efficiently processed by downstream steps of BER ().
The p300 deletion affects DNA repair of nicks- and nicks+mismatch-containing plasmids, but not damaged base-containing plasmids
Lysine acetyltransferase p300 is a major enzyme responsible for acetylation of histone and non-histone proteins. It is indicated as potentially responsible for acetylation of numerous factors involved in mechanisms of DNA repair and replication.Citation14,15,17,18,37-42 Consequently, we investigated whether the loss of p300 in cells would affect DNA repair of lesions in our plasmid constructs. The effect of the loss of p300 on DNA repair was analyzed in HCT116 and in p300 deficient cells (p300−/−) derived previously from HCT116 by targeting exon 2 of the EP300 gene.Citation6 HCT116 is hemizygous for p300 expressed from a single allele with a frameshift mutation in the last exon resulting in a truncated protein, but with an intact histone acetyltransferase domain. Both cell lines were transfected with undamaged plasmid (Control), nicks-containing plasmid (Nick) and a series of nicks+mismatch-containing plasmids, in which a mismatch was placed at various distances (2 to 30 nucleotides) from a nick located at their 3′ side ().
Compared to undamaged plasmid used as Control, the efficiency of mismatch repair in both cell lines decreased with increasing distance of the mismatch from the 5′ nick. This indicates that a repair process initiated at the nick was important for correction of the mismatch. Significantly, greater reductions in EGFP expression occurred at every distance for p300−/− cells, indicating that a lack of p300-guided acetylation affected repair of the lesions (). Mismatch repair at positions 13 and 23 was relatively robust in the wild-type (p300+) cells, suggesting that acetylation promotes replacement of a longer patch.
Figure 6. Effect of p300 depletion on repair of mismatches in cells with deficient MMR. (A) HCT116 and its p300 knockout derivative (HCT116 p300−/−) were transfected with undamaged plasmid (Control), nicks containing plasmid (Nick) and a series of nicks and mismatch containing plasmids. Stop codon-creating mismatches were placed at different distances from the 5′ nick. (B) Graph of relative expression levels of EGFP in HCT116, HCT116 p300−/− and rescue cells HCT116 p300−/−(p300+) expressing p300 from the plasmid. Relative expression levels represent the mean of triplicate experiments. (C) Representative contour plots of flow cytometric analysis of rescue in EGFP gene repair for the construct with nicks and series of constructs with nicks and a mismatch after transfection of p300 cDNA plasmid. The numbers indicate the percentages of GFP positive population. Median fluorescence intensities are indicated by a cross. All plots were gated on the living cells fraction.
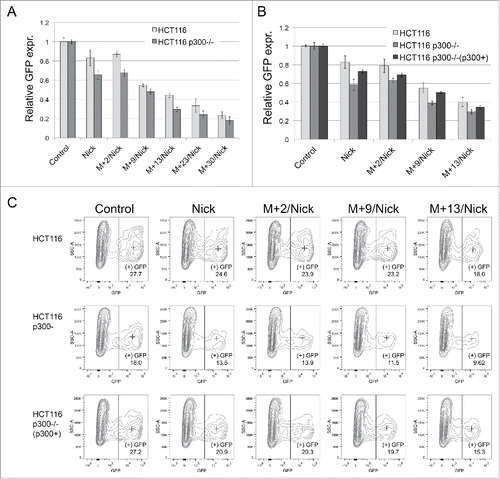
To determine whether the effect is specifically related to the loss of acetyltransferase, the p300 deficient cells were first transfected with plasmid containing EP300 cDNA, and 24 h later with Control or lesions-containing plasmids. For comparison, parallel transfections were performed with HCT116 and p300−/− cells, but with an empty plasmid followed by transfections with Control or lesions-containing plasmids. Flow cytometric analyses were performed 24 h after the second transfection. As shown in , the EGFP levels were partially restored for lesion-containing plasmids when p300 deficient cells were transfected with plasmid expressing p300. This result indicates that reductions in DNA repair observed in p300−/− cells are indeed caused by lack of the acetyltransferase.
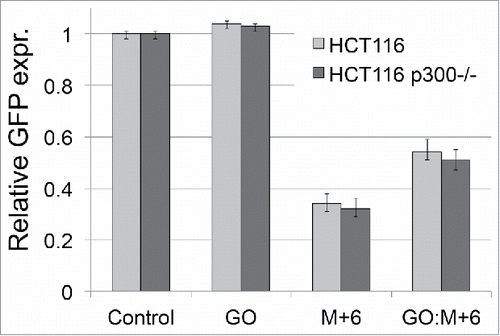
We also tested whether loss of p300 would affect BER mechanisms. p300 was previously indicated as a potential acetyltransferase able to modify BER pathway proteins, including Pol β, OGG1, RPA, PCNA and FEN1.Citation14,17,18,38,43 As shown in , no differences were observed in relative EGFP levels for the 8-oxoG containing plasmid (GO), as well as for GO:M+6, for both HCT116 cell lines. This result is inconsistent with previous reports showing that p300-mediated acetylation increased the efficiency of reconstituted LP-BER. Citation15 This specific outcome will be addressed in the Discussion.
Figure 7. Effect of p300 depletion on base excision repair. HCT116 and its p300 knockout derivative HCT116 p300−/− were transfected with undamaged plasmid (Control), plasmid with incorporated 8-oxoG (GO), and 2 plasmid constructs, M+6 and GO:M+6 used to monitor repair of 8-oxoG through the LP-BER pathway. Relative expression levels represent the mean of triplicate experiments performed in duplicate.
Discussion
Monitoring reactivation of expression of an EGFP gene bearing inactivating damage, present in a plasmid, is a convenient and effective approach for analyzing DNA repair mechanisms in cells. Such plasmids can be transfected into cell lines with modified or inactivated DNA repair proteins to assess the role of the proteins in repair of the inactivating damage. The goal of our studies was to develop such plasmid systems and use them to determine whether protein acetylation in cells influences DNA repair mechanisms, in particular MMR and BER.
In purified systems, nicks+mismatch-containing plasmids support nick-directed mismatch-provoked excision and repair.Citation44-46 In our cellular system, nicks guide the DNA repair proteins to work within only the transcribed strand of the EGFP gene, catalyzing repairs of the mismatch that are initiated from the nick located at its 5′ side. Two mechanisms may be used: the EXO1-dependent pathway involving excision of DNA fragment with the mismatch, followed by Pol δ - directed synthesis; or the EXO1-independent pathway proposed to involve synthesis-driven displacement of a DNA segment spanning the mismatch ().Citation24,26 HCT116 cells are deficient in MMR due to defective MLH1, which likely affects formation of the MMR complex and recruitment of EXO1.Citation24 In these cells, we expect that nicked DNA constructs with a mismatch become primary substrates for an EXO1-independent pathway ().Citation26
Previous studies showed that hyperacetylation induced by sodium butyrate stimulates nucleotide excision repair in UV-irradiated cells.Citation8,10,47-50 The stimulation of DNA repair was shown to be associated with alterations in chromatin structure making damaged sites in DNA more accessible to repair enzymes. Moreover, impaired chromatin relaxation observed in the presence of the HAT inhibitor garcinol, suppressed the NHEJ pathway in DSBs repair Citation36.
Acetylation and deacetylation have been shown, mostly through reconstitution reactions in vitro, to regulate the function of proteins involved in DNA replication and repair. Enzymes acetylated after purification displayed differences in catalytic properties from their un-acetylated counterparts. In our current work, we approached the experiments knowing that numerous DNA replication and repair proteins are acetylated by p300 and that acetylation changes their properties in a way that should promote more efficient strand displacement and consequently more effective repair.Citation15,17,18,43,51
In our studies, exposing cells to sodium butyrate (deacetylase inhibitor) produced an increase in repair of nicked plasmid DNA in HeLa cells, and also increases in repair of a mismatch and nicks in HeLa and MMR-deficient HCT116 cells. Exposing of cells to garcinol (acetyltransferase inhibitor) inhibited repair of the diagnostic nicks+mismatch-containing plasmid in both cell lines, and also repair of nicked DNA, but only at higher garcinol concentrations in HCT116 cells. The observed effects of sodium butyrate and garcinol on DNA repair in plasmid vectors indicate that MMR mechanisms are stimulated by acetylation. In HCT116 cells hyperacetylation had little effect on EGFP expression from nicks-containing plasmids, whereas increases of about 20% were observed for nicks+mismatch-containing plasmid.
Currently, it is known that acetylation and deacetylation of different K residues in MutS homolog 2 (MSH2) have positive effects on MMR mechanisms. MSH2 interacts with MSH6 to form the MutSα complex, which recognizes base-base mispairs and initiates the repair process. Acetylation of K residues at the C termini of MSH2 promoted formation of stable MutSα complexes. Citation19 Interestingly, other studies suggest that deacetylation of K residue on N termini of MSH2 promotes DNA MMR activity.Citation20 Acetylation was also detected in components of endonuclease MutLα, MLH1 and PMS2, as well as in EXO1.Citation52
Also, enzymes involved in Okazaki fragment maturation are influenced by acetylation to promote strand displacement synthesis.Citation16-18,53 Assessed in vitro, FEN1 had previously been reported to be inhibited approximately about 90% by acetylation.Citation18 This property would delay ligation of the fragments. We found that acetylation of the helicase/nuclease Dna2 is stimulated as much as 12-fold by acetylation.Citation16 This property would keep the displaced strand trimmed short, preventing formation of structures that interfere with displacement. We have additional preliminary evidence that the strand displacement synthesis functions of DNA polymerases δ and β are stimulated by acetylation (unpublished observation). These findings prompted us to propose that acetylation is employed by cells to increase the size of the patch that is removed from the 5′ end region of Okazaki fragments prior to ligation.Citation54 This size is significant because approximately 30 nucleotides of the 5′ region of each fragment are added by the 3′-exonuclease deficient, error-prone DNA polymerase α. If this patch is effectively removed, DNA replication becomes much more accurate. Recent localization of errors in the yeast and human genomes corresponded to synthesis by DNA polymerase α demonstrating that the error-prone patches are not always removed.Citation55 This suggests that protein acetylation is a likely regulator of fidelity during eukaryotic DNA replication.
The current understanding of eukaryotic DNA repair mechanisms indicates that the enzymes involved in DNA replication are also utilized in DNA repair.Citation56,57 In particular, the strand displacement synthesis described for Okazaki fragment maturation is also utilized in DNA repair processes such as mismatch repair, long patch base excision repair, and also nucleotide excision repair. Analyzing nicks+mismatch-containing plasmids, in which a mismatch was placed at various locations 3′ of a nick, we found that the efficiency of repairing the mismatch decreases with increasing distance from the nick. This indicates that repair of mismatches placed in various distances from a nick in our plasmid system depends primarily on a strand displacement synthesis initiated at the nick, also proposed in the EXO1-independent path model.Citation58 This is essentially the same strand displacement synthesis process, using the same protein components, as with Okazaki fragment processing. The negative effect of p300 loss on mismatch repair in this setting is consistent with results showing that acetylation improves strand displacement.Citation16-18,53 The implication of our results is that the acetylation-dependent increase in patch size observed for Okazaki fragment maturation in vitro, also occurs in cells. We conclude that the nick-dependent repair of mismatches and the 5′-end processing of Okazaki fragments are improved by protein acetylation-dependent increases in strand displacement synthesis. Consequently, this mechanism increases fidelity of both DNA replication and repair.
Surprisingly, from our analysis, the LP-BER mechanism in cells, which also involves strand displacement synthesis, was not influenced by acetylation. Whereas previous studies showed that LP-BER reactions in vitro were stimulated when performed with whole cells extracts prepared from cells exposed to sodium butyrate, we did not observe such stimulation in our cell-based system.Citation15 Also, p300-mediated acetylation of different factors involved in BER showed stimulation of synthesis-driven displacement utilized by LP-BER, whereas in our cell-based system depletion of p300 did not affect LP-BER.Citation15,17,18,43 Specifically, there was no evident effect of acetylation on repair of a mismatch downstream of an 8-oxoG damaged base (GO:MC6). However, the inhibitory effect of garcinol on repair in GO constructs suggests that at least some steps in BER are affected by loss of acetylation. Here, reduction in EGFP might result from accumulation of 1nt-gaps intermediates previously indicated to have inhibitory effect on transcription. We propose that the absence of an effect of acetylation on LP-BER results from overlapping of inhibitory and stimulatory effects on proteins involved in the early steps of BER, when the damaged base is removed.
The removal of 8-oxoG, leading to 1nt gap intermediates, can be done by different enzymatic activities (). For example, OGG1, in addition to excision of damaged base by β elimination, can perform strand cleavage using its AP lyase activity, which generates a 5′-phosphate and a 3′PUA (3′-phospho-α,β unsaturated aldehyde). The 3′PUA is subsequently removed by the AP endonuclease, APEX1. However, AP lyase activity of OGG1 is weak and strand cleavage is done with support of APEX1 generating 5′-dRP and 3′-OH. These ends can be processed by SP-BER, if the 5′dRP is removed by the 5′dRPase activity of Pol β, or by LP-BER, if 5′dRP is not removed. Alternatively, OGG1 can be removed from an AP site by NEIL1 protein, which generates 3′ and 5′ ends with phosphate residues.Citation59 These substrates will be only processed by SP-BER, which involves PNK and XRCC1.Citation60
In p300-mediated acetylation of OGG1, the glycosylase activity is stimulated but the abasic site binding of this enzyme is weakened. If OGG1 is replaced by APEX1, it will generate 5′-dRP and 3′-OH. Since acetylation of Pol β affects 5′dRPase activity, the repair is finished through LP-BER with support of PCNA and FEN1. However, acetylated OGG1 with reduced binding for AP site can also be removed by NEIL1, which binds abasic sites more strongly then OGG1.Citation59 Binding of NEIL1 will generate ends, which can only be processed through SP-BER. Consequently, whereas repair of the mismatch in GO:M+6 by the LP-BER mechanism might be stimulated by acetylation, the positive effect would be reduced if acetylated OGG1 is also replaced by NEIL1 leading to SP-BER, which would leave the mismatch unrepaired.
HCT116 is hemizygous for p300 expressed from a single allele with a frameshift mutation in the last exon resulting in a truncated protein. Although truncated p300 has an intact acetyltransferase domain, the BER mechanisms might be disrupted by the shorter version of this protein, for example, by dominant negative interactions. Additionally, while many studies have implicated p300 in acetylating DNA repair enzymes, we cannot rule out redundant functions by other acetyltransferease that may compensate for the loss of p300 in this cell line. We conclude that BER is a complex pathway that is regulated both positively and negatively by acetylation.
The effects of acetylation reported here are almost certainly not resulting from activation of chromatin. A plasmid system such as the one we used is not expected to form obstructive chromatin during the course of the assay.Citation32 Moreover, our damaged EGFP expression results are always presented with respect to the expression level of an undamaged EGFP control in the same plasmid structure environment. Additionally, different types of damage result in different levels of expression. For example, garcinol had little effect on expression from nicked plasmids but caused a distinct reduction of expression from the nick + mismatch plasmids. The expression values with sodium butyrate had a distinctly different profile. When 8-oxo-G plasmids were used, the expression profile was also much different. If all of these measurements were just responding to a general change in chromatin accessibility, we would have expected more consistency in the response. As such, our results are also consistent with biochemical findings.
Overall, our plasmid systems represent the most effective way available to assess the effects of protein acetylation on mammalian DNA replication and repair. Moreover, our results strongly indicate that protein acetylation alters mammalian DNA replication and repair pathways to improve their efficiency and fidelity.
Material and methods
Materials
Wild-type HCT116 cells were purchased from Sigma. The HCT116 p300 knockout D10 clone was purchased from Cancer Research UK Cambridge Institute, pCMV-kEGFPluc plasmid was generously provided by Dr Patrick Calsou and plasmid pcDNA3.1-p300 containing EP300 cDNA was a gift from Warner Greene (Addgene plasmid # 23252). Synthetic unmodified and 8-oxoG-containing oligonucleotides (indicated in ) were purchased from Eurofins MWG Operon and Integrated DNA Technologies.
Preparation of double-nicked plasmids
Plasmid constructs used in our cell-based system were based on pCMV-kEGFPluc, containing N-terminal-modified EGFP-luciferase fusion protein under the control of a CMV promoter.Citation27 In order to efficiently perform site-specific incorporation of 8-oxoG and/or mismatches into the EGFP open reading frame, we constructed two new plasmids, pD59 and pD61, built on the pCMV-kEGFPluc vector backbone, into which we introduced 2 restriction sites recognized by the sequence-specific nicking enzyme Nb.BbvCI.Citation30 One site was placed just upstream of the EGFP ORF and the second site was placed within the ORF about 30nt and 35nt downstream in pD59 and pD61, respectively (). The bottom DNA strand of the gene (“top” refers to the protein-coding strand or untranscribed strand, and “bottom” refers to the transcribed strand) was selectively nicked by incubating 60 μg of plasmid DNA with 13U of Nb.BbvCI (New England BioLabs) for 1.5 h at 37°C in 120 μl of CutSmart Buffer (supplied by the manufacturer). The enzyme was heat inactivated (80°C, 25 min), and the presence of 2 nicks in the same DNA strand was verified as follows. Nicked plasmid (1μg of DNA) was supplemented with 90 pmol of synthetic oligonucleotide and adjusted to 10 μl of T4 DNA ligase buffer (Thermo Scientific). The mixture was incubated at 37°C for 30 min in the presence of PNK and then incubated at 80°C for 10 min in a thermal cycler, followed by slow cooling to 50°C at a temperature transition rate of −0.02°C × s−1. Subsequently, the samples were rapidly chilled to 4°C, then 1 Weiss U of T4 DNA ligase (Thermo Scientific) was added, and samples were incubated for 18 h at 16°C. Parallel reactions were set without PNK, to avoid 5′ end phosphorylation of competitor oligonucleotides, to serve as controls of efficient replacement of original single stranded DNA by synthetic oligonucleotide. Samples were analyzed on a 0.8% agarose gel containing 0.5 mg/L ethidium bromide. Complete inhibition of ligation in the reactions incubated with the nonphosphorylated competitor oligonucleotide indicated the presence of 2 nicks in the same DNA strand and replacement original ssDNA fragment with synthetic oligonucleotide, which was a prerequisite for the subsequent procedures.
Preparation of supercoiled plasmids with inserted synthetic oligonucleotides into the protein-coding region of the EGFP gene
The supercoiled plasmids (CCC forms) with inserted synthetic oligonucleotides were prepared from the double-nicked plasmid substrates as follows. Double-nicked plasmid (30 μg of DNA) was supplemented with 1.36 nmol of synthetic oligonucleotide and adjusted to 300 μl with T4 DNA ligase buffer (Thermo Scientific), or scaled up appropriately. The mixture was incubated at 37°C for 30 min in the presence of 150 U of PNK. The excised 39-mers were melted out (80°C, 10 min) and slowly cooled to 50°C at a temperature transition rate of −0.02°C × s−1 to allow annealing of the 39-mers with the double-nicked plasmid. Next, the samples were rapidly chilled to 4°C, and ligation reactions were performed for 18 h at 16°C in the presence of 30 Weiss U of T4 DNA ligase. Closed circular DNA was purified by cesium chloride density centrifugation.
Preparation of gapped plasmids
Gapped plasmids were prepared from the double-nicked plasmid substrates as above, except after annealing the reactions were not subjected to ligation and cesium chloride density gradient purification.
Cell culture, transfections and analysis of GFP expression
HeLa cells were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 1% penicillin/streptomycin (Invitrogen). HCT116 and HCT116 p300−/− were cultured in McCoy's 5A medium supplemented with 10% FBS, 2 mM glutamine, and 1% penicillin/streptomycin. For transfection experiments, 1.5 × 105 cells (in 2 ml) were seeded into 6-well dishes, and 24 h later they were transfected with plasmid constructs (1 μg) using Lipofectamine 3000 (HeLa) or Lipofectamine LTX Plus (HCT116) according to the manufacturer's protocol (Invitrogen). To ensure that cells first responded to inhibitors of acetyltransferase and deacetylase, before performing DNA repairs on plasmids, fresh media with and without garcinol and sodium butyrate (at indicated concentrations) were added 3–6 h before transfection. 18 h after transfection cells were washed with PBS and recovered by scraping in PBS buffer with 15% FBS. From each transfection, 20,000 cells were counted and their green fluorescence intensity was measured using an LSR II 18-Color flow cytometer (Becton Dickinson). Relative EGFP expression (RE) was calculated with the following formula RE = (M · IM)/(C · IC); where M and C are the percentages of green cells transfected with damaged plasmid and undamaged control plasmid, respectively. IM and IC are the mean fluorescence intensities of EGFP positive cells for M and C, respectively.Citation25
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We are grateful to Dr Patrick Calsou (Institute of Pharmacology and Structural Biology, CNRS UMR) for pCMV-kEGFPluc used in these studies. We are also grateful to Dr Warner Greene (University of California, San Francisco) for providing pcDNA3.1-p300 plasmid.
Funding
This work was supported by the National Institutes of Health [CA176249 to R.A.B. and GM098328 to L.B.]. Funding for open access charge: National Institutes of Health.
References
- Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41:185-98; PMID:18804549; http://dx.doi.org/10.1016/j.biocel.2008.08.027
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15-23; PMID:16289629; http://dx.doi.org/10.1016/j.gene.2005.09.010
- Sadoul K, Wang J, Diagouraga B, Khochbin S. The tale of protein lysine acetylation in the cytoplasm. J Biomed Biotechnol 2011; 2011:970382; PMID:21151618; http://dx.doi.org/10.1155/2011/970382
- Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell Mol Life Sci 2001; 58:728-36; PMID:11437234; http://dx.doi.org/10.1007/PL00000896
- Xu W, Li Y, Liu C, Zhao S. Protein lysine acetylation guards metabolic homeostasis to fight against cancer. Oncogene 2014; 33:2279-85; PMID:23665675; http://dx.doi.org/10.1038/onc.2013.163
- Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, Brindle K, Aparicio S, Caldas C. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci U S A 2004; 101:7386-91; PMID:15123817; http://dx.doi.org/10.1073/pnas.0401002101
- Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A 2012; 109:10843-8; PMID:22711838; http://dx.doi.org/10.1073/pnas.1201487109
- Dresler SL. Stimulation of deoxyribonucleic acid excision repair in human fibroblasts pretreated with sodium butyrate. Biochemistry 1985; 24:6861-9; PMID:4074730; http://dx.doi.org/10.1021/bi00345a019
- Smith PJ. n-Butyrate alters chromatin accessibility to DNA repair enzymes. Carcinogenesis 1986; 7:423-9; PMID:3753905; http://dx.doi.org/10.1093/carcin/7.3.423
- Smerdon MJ, Lan SY, Calza RE, Reeves R. Sodium butyrate stimulates DNA repair in UV-irradiated normal and xeroderma pigmentosum human fibroblasts. J Biol Chem 1982; 257:13441-7; PMID:7142158
- Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002; 419:411-5; PMID:12353039; http://dx.doi.org/10.1038/nature01035
- Megee PC, Morgan BA, Smith MM. Histone H4 and the maintenance of genome integrity. Genes Dev 1995; 9:1716-27; PMID:7622036; http://dx.doi.org/10.1101/gad.9.14.1716
- Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 2006; 16:433-42; PMID:16904321; http://dx.doi.org/10.1016/j.tcb.2006.07.007
- Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Burki C, Imhof R, Schar P, Hottiger MO. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol Cell 2002; 10:1213-22; PMID:12453427; http://dx.doi.org/10.1016/S1097-2765(02)00745-1
- Muftuoglu M, Kusumoto R, Speina E, Beck G, Cheng WH, Bohr VA. Acetylation regulates WRN catalytic activities and affects base excision DNA repair. PLoS One 2008; 3:e1918; PMID:18398454; http://dx.doi.org/10.1371/journal.pone.0001918
- Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J Biol Chem 2010; 285:4398-404; PMID:20019387; http://dx.doi.org/10.1074/jbc.M109.086397
- Naryzhny SN, Lee H. The post-translational modifications of proliferating cell nuclear antigen: acetylation, not phosphorylation, plays an important role in the regulation of its function. J Biol Chem 2004; 279:20194-9; PMID:14988403; http://dx.doi.org/10.1074/jbc.M312850200
- Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell 2001; 7:1221-31; PMID:11430825; http://dx.doi.org/10.1016/S1097-2765(01)00272-6
- Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Mol Cell 2014; 55:31-46; PMID:24882211; http://dx.doi.org/10.1016/j.molcel.2014.04.028
- Radhakrishnan R, Li Y, Xiang S, Yuan F, Yuan Z, Telles E, Fang J, Coppola D, Shibata D, Lane WS, et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J Biol Chem 2015; 290:22795-804; PMID:26221039; http://dx.doi.org/10.1074/jbc.M114.612945
- Goellner EM, Smith CE, Campbell CS, Hombauer H, Desai A, Putnam CD, Kolodner RD. PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell 2014; 55:291-304; PMID:24981171; http://dx.doi.org/10.1016/j.molcel.2014.04.034
- Smith CE, Bowen N, Graham WJt, Goellner EM, Srivatsan A, Kolodner RD. Activation of saccharomyces cerevisiae Mlh1-Pms1 endonuclease in a reconstituted mismatch repair system. J Biol Chem 2015; 290:21580-90; PMID:26170454; http://dx.doi.org/10.1074/jbc.M115.662189
- Desai A, Gerson S. Exo1 independent DNA mismatch repair involves multiple compensatory nucleases. DNA Repair (Amst) 2014; 21:55-64; PMID:25037770; http://dx.doi.org/10.1016/j.dnarep.2014.06.005
- Goellner EM, Putnam CD, Kolodner RD. Exonuclease 1-dependent and independent mismatch repair. DNA Repair (Amst) 2015; 32:24-32; PMID:25956862; http://dx.doi.org/10.1016/j.dnarep.2015.04.010
- Lei X, Zhu Y, Tomkinson A, Sun L. Measurement of DNA mismatch repair activity in live cells. Nucleic Acids Res 2004; 32:e100; PMID:15249596; http://dx.doi.org/10.1093/nar/gnh098
- Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, Modrich P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci U S A 2009; 106:8495-500; PMID:19420220; http://dx.doi.org/10.1073/pnas.0903654106
- Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep 2003; 4:363-7; PMID:12671676; http://dx.doi.org/10.1038/sj.embor.embor796
- Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 2004; 3:483-94; PMID:15084310; http://dx.doi.org/10.1016/j.dnarep.2004.01.003
- Kathe SD, Shen GP, Wallace SS. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J Biol Chem 2004; 279:18511-20; PMID:14978042; http://dx.doi.org/10.1074/jbc.M313598200
- Kitsera N, Stathis D, Luhnsdorf B, Muller H, Carell T, Epe B, Khobta A. 8-Oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1. Nucleic Acids Res 2011; 39:5926-34; PMID:21441539; http://dx.doi.org/10.1093/nar/gkr163
- Brand M, Moggs JG, Oulad-Abdelghani M, Lejeune F, Dilworth FJ, Stevenin J, Almouzni G, Tora L. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 2001; 20:3187-96; PMID:11406595; http://dx.doi.org/10.1093/emboj/20.12.3187
- Jeong S, Stein A. Micrococcal nuclease digestion of nuclei reveals extended nucleosome ladders having anomalous DNA lengths for chromatin assembled on non-replicating plasmids in transfected cells. Nucleic Acids Res 1994; 22:370-5; PMID:7510391; http://dx.doi.org/10.1093/nar/22.3.370
- Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A 2010; 107:16066-71; PMID:20713735; http://dx.doi.org/10.1073/pnas.1010662107
- Dherin C, Gueneau E, Francin M, Nunez M, Miron S, Liberti SE, Rasmussen LJ, Zinn-Justin S, Gilquin B, Charbonnier JB, et al. Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Mol Cell Biol 2009; 29:907-18; PMID:19015241; http://dx.doi.org/10.1128/MCB.00945-08
- Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, Kundu TK. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem 2004; 279:33716-26; PMID:15155757; http://dx.doi.org/10.1074/jbc.M402839200
- Oike T, Ogiwara H, Torikai K, Nakano T, Yokota J, Kohno T. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int J Radiat Oncol Biol Phys 2012; 84:815-21; PMID:22417805; http://dx.doi.org/10.1016/j.ijrobp.2012.01.017
- Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res 2004; 32:3033-9; PMID:15175427; http://dx.doi.org/10.1093/nar/gkh632
- Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol 2006; 26:1654-65; PMID:16478987; http://dx.doi.org/10.1128/MCB.26.5.1654-1665.2006
- Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 2001; 410:387-91; PMID:11268218; http://dx.doi.org/10.1038/35066610
- Cazzalini O, Sommatis S, Tillhon M, Dutto I, Bachi A, Rapp A, Nardo T, Scovassi AI, Necchi D, Cardoso MC, et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res 2014; 42:8433-48; PMID:24939902; http://dx.doi.org/10.1093/nar/gku533
- Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, Scovassi AI, Prosperi E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst) 2012; 11:844-52; PMID:22954786; http://dx.doi.org/10.1016/j.dnarep.2012.08.001
- Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell 2002; 9:265-77; PMID:11864601; http://dx.doi.org/10.1016/S1097-2765(02)00453-7
- Wu Y, Chen H, Lu J, Zhang M, Zhang R, Duan T, Wang X, Huang J, Kang T. Acetylation-dependent function of human single-stranded DNA binding protein 1. Nucleic Acids Res 2015; 43:7878-87; PMID:26170237; http://dx.doi.org/10.1093/nar/gkv707
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell 2006; 126:297-308; PMID:16873062; http://dx.doi.org/10.1016/j.cell.2006.05.039
- Genschel J, Modrich P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell 2003; 12:1077-86; PMID:14636568; http://dx.doi.org/10.1016/S1097-2765(03)00428-3
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5'-directed human mismatch repair in a purified system. Cell 2005; 122:693-705; PMID:16143102; http://dx.doi.org/10.1016/j.cell.2005.06.027
- Ramanathan B, Smerdon MJ. Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J Biol Chem 1989; 264:11026-34; PMID:2738057.
- Krishnan V, Liu B, Zhou Z. 'Relax and Repair' to restrain aging. Aging (Albany NY) 2011; 3:943-54; PMID:22067432; http://dx.doi.org/10.18632/aging.100399
- Zhang W, Wu H, Yang M, Ye S, Li L, Zhang H, Hu J, Wang X, Xu J, Liang A. SIRT1 inhibition impairs non-homologous end joining DNA damage repair by increasing Ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 2015 Dec 3; [Epub ahead of print]; PMID:26646449; http://dx.doi.org/10.18632/oncotarget.6455
- Johnson DP, Spitz GS, Tharkar S, Quayle SN, Shearstone JR, Jones S, McDowell ME, Wellman H, Tyler JK, Cairns BR, et al. HDAC1,2 inhibition impairs EZH2- and BBAP-mediated DNA repair to overcome chemoresistance in EZH2 gain-of-function mutant diffuse large B-cell lymphoma. Oncotarget 2015; 6:4863-87; PMID:25605023; http://dx.doi.org/10.18632/oncotarget.3120
- Balakrishnan L, Bambara RA. Okazaki fragment metabolism. Cold Spring Harb Perspect Biol 2013; 5:a010173; PMID:23378587; http://dx.doi.org/10.1101/cshperspect.a010173
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834-40; PMID:19608861; http://dx.doi.org/10.1126/science.1175371
- Balakrishnan L, Bambara RA. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J Biol Chem 2011; 286:6865-70; PMID:21177245; http://dx.doi.org/10.1074/jbc.R110.209502
- Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem 2013; 82:119-38; PMID:23451868; http://dx.doi.org/10.1146/annurev-biochem-072511-122603
- Reijns MA, Kemp H, Ding J, de Proce SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature 2015; 518:502-6; PMID:25624100; http://dx.doi.org/10.1038/nature14183
- Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 2011; 147:1040-53; PMID:22118461; http://dx.doi.org/10.1016/j.cell.2011.10.025
- Masih PJ, Kunnev D, Melendy T. Mismatch Repair proteins are recruited to replicating DNA through interaction with Proliferating Cell Nuclear Antigen (PCNA). Nucleic Acids Res 2008; 36:67-75; PMID:17984070; http://dx.doi.org/10.1093/nar/gkm943
- Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem 2005; 280:39752-61; PMID:16188885; http://dx.doi.org/10.1074/jbc.M509701200
- Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry 2004; 43:11596-604; PMID:15350146; http://dx.doi.org/10.1021/bi049097i
- Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 2008; 18:27-47; PMID:18166975; http://dx.doi.org/10.1038/cr.2008.8