
ABSTRACT
Purpose: The HMGI-C (high mobility group protein isoform I-C) protein is a member of the high-mobility group AT-hook (HMGA) family of small non-histone chromosomal proteins that can modulate transcription of an ample number of genes. Genome-wide studies reveal upregulation of the HMGI-C gene in many human cancers, which suggests that HMGI-C might play a critical role in the progression of various tumors. However, the exact role of HMGI-C in breast adenocarcinoma has not been made clear. Methods: HMGI-C mRNA expression in breast cancer samples and marginal normal tissues was characterized using qRT-PCR. The cytotoxic effects of HMGI-C siRNA on breast adenocarcinoma cells were determined using MTT assay. Relative HMGI-C mRNA and protein levels were measured by quantitative real-time PCR and western blotting, respectively. Apoptosis detection was done using TUNEL and Annexin-V/PI assays, P53, caspase 3, 9, 8 and Bcl2 proteins evaluated by protein gel blot and miR34a, Let-7a genes investigates by QRT-PCR assay. Cell cycle was analyzed by flow cytometry assay using propidium iodide DNA staining. Results: An overexpression of HMGA2 was revealed with highly statistically significant differences between breast cancer samples and marginal normal tissues (P < 0.0001). HMGI-C siRNA significantly reduced both mRNA and protein expression levels in a 48-hour period after transfection and in a dose-dependent manner. We observed that the knockdown of HMGI-C led to the significant induction of apoptosis via mitochondrial pathway by inducing miR34a and cell cycle arrest in MDA-MB-468 cells in vitro. Conclusions: These results propose that HMGI-C might play a critical role in the progression of breast adenocarcinoma. Here we introduced HMGI-C as a potential therapeutic target for trigger apoptosis and cell cycle arrest in human breast adenocarcinoma. Therefore HMGI-C siRNA may be an effective adjuvant in human breast adenocarcinoma.
Introduction
Breast cancer is the most common cancer diagnosed among women worldwide, accounting for nearly 1 in 3 cancers, and it is the second cause of cancer death among women, following lung cancer.Citation1 Despite the great improvements in clinical and therapeutic techniques in recent years, many advanced breast cancer patients still die of postoperative recurrence and metastasis disease. One of the primary reasons for ineffective therapies for these patients is our lack of understanding about the complete and accurate molecular mechanisms involved in carcinogenesis, progression and invasion of breast cancer. Therefore, the innovation of new treatment modalities to overcome these ineffective therapies of tumor cells may be a potential source of improved therapies.
Using small interfering RNA (siRNA) has been a strategy for studying gene function and disease treatment. There are commonly multiple RNAi gene therapy strategies being studied clinically for the treatment of respiratory syncytial virus infection and age-related macular degeneration (AMD).Citation2 The effectiveness of siRNA in cancer treatment is allocated because of its potential and high efficiency, knock downing in the advanced stages of growth and costing less than other methods of gene therapy,Citation3-5 and offering high specificity compared to other cancer therapy methods such as operation and chemotherapy.Citation6,7
HMGI-C protein, also known as HMGA2 protein, belongs to the family of nuclear non-histone phosphoproteins called the high mobility group (HMGA). These proteins contain 3 basic short sequences, called the AT-hook. These basic sequences bind to AT-rich regions of the minor groove of B-form DNA. These proteins are involved in many fundamental cellular processes, including mitosis, cell-cycle control, cell division, regulation of transcription (by binding to transcription factors such as NF-kB, ATF-2/c-Jun, Elf-1, Oct-2, Oct-6, SRF, NF-Y, PU-1, RAR), differentiation and cellular aging.Citation8-10 HMGI-C protein is relatively overexpressed where cells proliferate rapidly (early embryo tissues), parenchymal organs, proliferating epithelial cells,Citation11 mesenchymal cell condensations and mesenchymal derivatives.Citation12 The expression of HMGA genes are suppressed in differentiated cells and the HMGI-C gene is underexpressed in adult human tissues other than embryonic tissues.Citation13,14 In contrast HMGI-C gene overexpression is observed in many human malignancies such as non-small lung cancers,Citation15 pancreatic carcinoma,Citation16 epithelial ovarian cancers,Citation17 colorectal cancer,Citation18 retinoblastomas,Citation19 squamous cell carcinomas,Citation20 myeloproliferative disorderCitation21 and it has also been found to participate in EMT.Citation22,23 The studies related to breast cancer suggest that transcriptional re-expression of HMGI-C may be important in the progression of breast cancer.
Recent studies described the mechanism of post-transcription control of the HMGI-C gene expression by microRNA.Citation24-26 Let-7 was described as a negative regulator of the HMGI-C gene. Recently, microRNAs were shown to regulate the expression of an ample amount of genes, especially tumor suppressor genes and oncogenes including p53 and Bcl2.Citation27 However, despite the extensive research done in the field of microRNAs, the functional role of microRNAs is not completely understood. Many of the miRNAs play a role in the regulation of apoptosis. Recent studies have shown the effect of apoptosis-related gene alternation in miRNA change during cancer therapy. For instance, the most significant induction is related to miR-34a and p53, which have a direct effect on the regulation of each other.Citation28,29
In this study, we show a valid cancer-relevant overexpression of the HMGI-C level that is associated with the survival of the human breast adenocarcinoma cell (MDA-MB-468). We used a small interference RNA technique to suppress HMGI-C expression in the human breast adenocarcinoma MDA-MB-468 cell line and investigated the effect of HMGI-C silencing on apoptosis and the cell cycle. Remarkably, here we report that HMGI-C is a direct transcriptional target of miR-34a. We demonstrate that suppression of HMGI-C promotes apoptosis and leads to dramatic global alterations in miR-34a expression.
Results
HMGI-C overexpression in breast cancer tissues and cultured cells
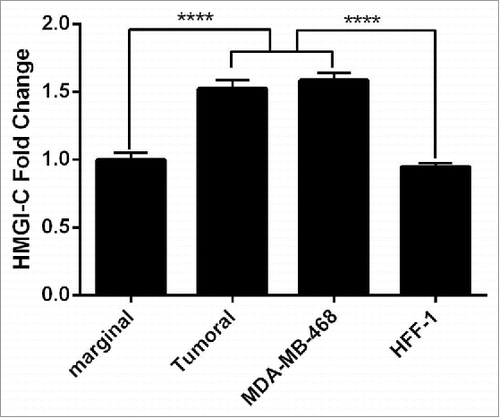
In this study, we evaluated the expression level of HMGI-C genes in 15 breast cancer samples and 15 marginal normal tissues, using quantitative real time PCR. The expression levels of the HMGI-C gene were detected in all samples of breast cancer tissues versus marginal normal tissues (). In addition, the results of HMGI-C expression in breast adenocarcinoma cells (MDA-MB468) and normal fibroblast cells (HFF-1) showed the significant overexpression of HMGI-C in MDA-MB-468 cells compared to HFF-1 cells ().
Figure 1. Analysis of the HMGI-(C)mRNA expression in breast cancer patients. HMGI-C mRNA expression levels evaluated in 12 marginal tissues, 12 tumoral tissues, MDA-MB-468 and HFF-1 cells. ****p < 0.0001 vs. marginal tissues group and HFF-1 cells.
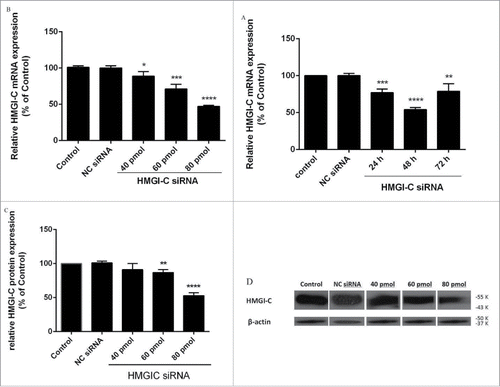
siRNA suppressed HMGI-C mRNA and protein levels in breast adenocarcinoma cells
First, we explored the effect of siRNA on HMGI-C gene expression in MDA-MB468 cells by qRT-PCR and western blot analysis. Relative HMGI-C gene expression was calculated in relation to the control group, which was considered 100%. As shown in , HMGI-C siRNA led to a marked time-dependent reduction of HMGI-C mRNA and both dose-dependent mRNA and protein levels (p < 0.05; relative to the control). At 24, 48 and 72 h after the transfection, the relative HMGI-C mRNA expression levels were 77.06%, 54.10% and 78.95%, respectively (), and the dose-dependent at 40, 60, 80 pmol of HMGI-C siRNA transfection relative HMGI-C mRNA expression levels were 88.87%, 71.13% and 47.25%, respectively (). HMGI-C protein expression levels were 67.10%, 11.65% and 8.20%, respectively () (p < 0.05). Notably, treatment with NC siRNA had a minimal effect on mRNA and protein levels compared with the control group. After the time and dose optimization, the best knockdown was achieved in 48 h and 80 pmol siRNA, further experiments were performed in the same conditions.
Figure 2. Suppression of HMGI-(C)mRNA and protein expression by siRNA in breast adenocarcinoma. HMGI-C mRNA expression in MDA-MB-468 cells, which were transfected with specific siRNA or (NC) siRNA after (A) 24, 48, 72 h, and (B) 40 pmol, 60 pmol, 80 pmol of specific siRNA. Relative HMGI-C mRNA expression was measured by qRT-PCR using 2(-ΔΔCt) method. (C) A representative protein gel blot of b-actin and HMGI-C proteins from cells transfected with HMGI-C siRNA or (NC) siRNA. (D) The expression level of each band was quantified using densitometry and normalized to the respective B-actin. The results were expressed as mean ± SD (n = 3); *p < 0.05, **p < 0.001, *** p = 0.0001, ****p < 0.0001 versus control group.
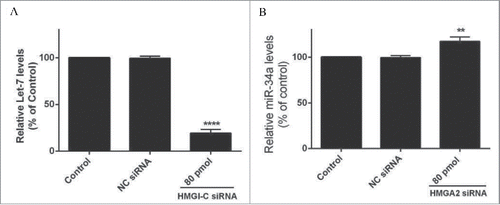
HMGI-C silencing cause down regulation in Let-7 and induction in miR-34a levels
A QRT-PCR analysis was performed for evaluation of the Let-7, miR-34a expression after HMGI-C knock downing by specific siRNA. The results showed Let-7 and miR-34a expression was calculated in relation to the control group. As shown in , HMGI-C silencing caused downregulation of Let-7 and induction of miR-34a in the treated group compared to the control group (p < 0.05). The relative Let-7 expression levels in the treated group were reduced to 19.33% and miR-34a expression increased to 117.28% ().
Figure 3. Relative Let-7a (A) and miR-34a (B) expression levels were measured by qRT-PCR using 2(-ΔΔCt) method. **p < 0.001, ****p < 0.0001 vs. control group.
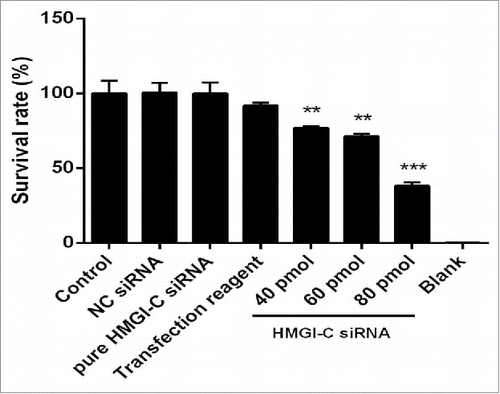
HMGI-C suppression caused reduction on breast adenocarcinoma cell survivability
To assess whether downregulation of HMGI-C has a cytotoxicity effect in breast adenocarcinoma cells, a treatment of HMGI-C siRNA or NC siRNA was performed on MDA-MB468 cells. As shown in , treatment with HMGI-C siRNA induced cytotoxicity in a dose-dependent way. The results of MTT assay showed that the IC50 of HMGI-C siRNA is 65.73 pmol and transfection interfere materials are not significantly toxic for the cells (p < 0.05). In contrast, no significant differences in cell viability were found between the NC siRNA transfected cells and the control group (p < 0.05) ().
Figure 4. Effect of HMGI-C siRNA on the MDA-MB-468 cells. 48 h after transfection with HMGI-C siRNA (40, 60, 80 pmol), cytotoxicity of treatments was determined by MTT assay. The results were expressed as mean ± SD (n = 3); **P < 0.01, ***P < 0.001 versus control group.
Suppression of HMGI-C induced apoptosis through intrinsic pathway
To understand the sensitizing effect of HMGI-C siRNA that was linked to the enhancement of apoptosis, the HMGI-C siRNA was transfected into MDA-MB-468 cells. The number of surviving cells was decreased compared to the control group. We found the amount of this decrease was impressive, because the transfection efficiency for the overall population was more than 40%. The depression in a tumor cell number was associated with the appearance of TUNEL-positive cells (). To investigate whether cell death was induced by specific siRNA through apoptosis, Annexin V/PI assay was performed. As shown in , we saw an increase in the cell population stained with Annexin V. We did not see an obvious change in the cell population stained with PI. In both TUNEL and Annexin V/PI assay, treatment with NC siRNA displayed no distinct alterations in the extent of apoptosis relative to the control group (p>0.05).
Figure 5. siRNA-mediated targeting of HMGI-C strongly sensitized MDA-MB-468 cells by stimulation of apoptosis. Untreated (A), and 80 pmol HMGI-C treated MDA-MB-468 (B) were prepared and assayed with TUNEL assay. FACS analysis of cell distribution for apoptosis and necrosis on MDA-MB-468 cells for untreated (C), (NC) siRNA (D) and 80 pmol siRNA (E), prepared after 48 h. Percentage of dual-positive (Annexin V and PI positive) cells from 3 independent experiments were quantified and presented as mean ± SD (n = 3). (F) Relative P53, caspase3, 9, 8, Bcl2 (5G, 5H) protein expression were evaluated by immune-blotting and densitometry using imageJ software. *p < 0.05, ****p < 0.0001, vs. control group.
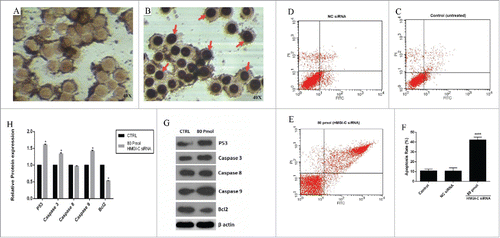
To determine which apoptosis pathway was involved, the levels of P53, caspase-3 and caspase-9 protein expression in 80 pmol treated group were significantly higher than those controls. In contrast, the levels of Bcl2 protein in the treated group were significantly lower than controls. Interestingly, we did not see a significant difference between the caspase 8 levels in those groups ().
HMGI-C mediated silencing arrest G0/G1 in breast adenocarcinoma cells
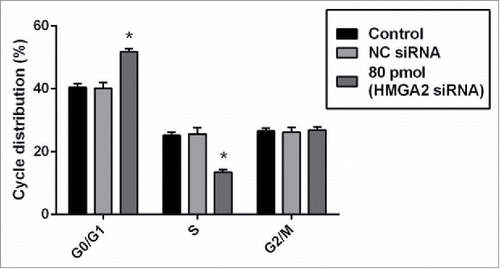
To observe whether the sensitizing effect of HMGI-C siRNA was linked to the arresting cell cycle or not, we did FACS analysis. When the HMGI-C siRNA was transfected into MDA-MB-468 cells, HMGI-C knock downing prevented mitotic entry by inducing G0/G1 arrest and eliminating cells with impaired DNA. We performed FACS analysis to determine the effect of HMGI-C knock downing on cell cycle distribution. FACS cell cycle profiles revealed a gradual transition from G0/G1 to S phase in transfected HMGI-C cells during 48 h incubation after transfection of 80 pmol specific HMGI-C siRNA. At 48 h incubation, 51.71% of the MDA-MB-468 cells were in G0/G1, 13.12% in S and 29.16 % in G2/M while NC siRNA displayed no significant difference (p > 0.05) ().
Figure 6. siRNA-mediated targeting of HMGI-C, prolonged G0/G1 arrest in MDA-MB-468 cells. Untreated, (NC) siRNA and 80 pmol HMGI-C treated cells were prepared and stained with the propidium iodide (PI). The percentage of the cell population in untreated, negative-control siRNA and 80 pmol HMGI-C siRNA group evaluated in each phases. The results are expressed as mean ± SD (n = 3); *p < 0.05, versus control group.
Discussion
Overexpression of oncogenes causes tumor progression in many human cancers. Some of the oncogenes,Citation30,31 have the ability to regulate not only proliferation but also inhibit apoptosis in many kinds of cancers including breast adenocarcinoma.Citation32 Previous studies also have proved that HMGI-C plays an important role in the processes of many kinds of human malignant epithelial tumors, including the functions of enhancing cell growth and invasion. Recently, the clinical association of HMGI-C in breast cancer has been reported.Citation33,34 The data show HMGI-C is overexpressed in human breast adenocarcinoma, and might be involved in apoptosis and the cell cycle. However, the essential roles of HMGI-C in cancer remain unclear.
To clarify the role of HMGI-C in cancer, especially in breast adenocarcinoma, HMGI-C expression was suppressed by specific siRNA in breast adenocarcinoma cells. Cytotoxicity assay showed the IC50 of specific HMGI-C siRNA in concentration of 65.73 pmol. The data showed HMGI-C may play a critical role in the survival and growth of cancerous cells.
An apoptosis assay showed the cell death was applied by means of apoptosis after blocking the expression of HMGI-C.Citation35,36 The results of TUNEL and Annexin V/PI assays showed, downregulation of HMGI-C expression significantly induced apoptosis in MDA-MB-468 cells. Furthermore, overexpression of the apoptosis-related proteins including P53, caspase 3 and caspase 9, and reduction of anti-apoptotic Bcl2 confirm the apoptosis induction strongly. This suggests that the major pathway for cell death through HMGI-C inhibition was a mitochondrial pathway.
To see the impact of micro-RNA involvement in the activation of apoptosis, we measured the expression levels of the miR-34a and Let-7 in HMGI-C silenced cells. The results suggested miR-34a induction is related to HMGI-C silencing. This finding revealed the probable roles of miR-34a induction on overexpression of P53 and downregulation of Bcl2 in the apoptosis pathway.Citation37,38 Let-7a is known as HMGI-C regulator,Citation22 and interestingly, the Let-7a expression level was downregulated followed by HMGI-C silencing. The result suggested that by knock downing the HMGI-C, the necessity for the presence of the Let-7a was lost.
We also assessed the cell cycle changes of untransfected NC siRNA and transfected MBA-MB-468 cells, and the results indicated suppression of HMGI-C expression could arrest the G0/G1 phase of the cell cycle in MDA-MB-468 cells significantly.
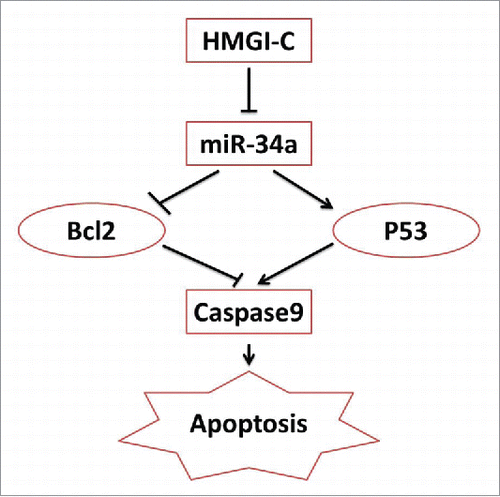
These data provide evidence that HMGI-C may be an important regulator of human breast adenocarcinoma as an apoptosis inhibitor and a cell cycle inducer. Our results are similar to those of previous studies on other kinds of cancersCitation17,35,39,40 and we suggested the new anti-apoptotic signaling axis model through HMGI-C and miR-34a ().
In this model, HMGI-C induced an anti-apoptotic pathway by inhibiting miR-34a. The results suggest miR-34a expression levels correlate with the apoptosis. miR-34a activates the tumor suppressor p53, and downregulates Bcl2. Following mir-34a/P53, caspase 9 was active through mitochondrial pathway and apoptosis happened ().Citation38
The research team concluded that HMGI-C targeting might represent a promising therapeutic approach for activating the apoptosis and cell division inhibition in breast adenocarcinoma patients with HMGI-C overexpression. Furthermore, additional in vitro and in vivo studies are needed to clarify the roles of HMGI-C in human cancers.
Materials and methods
Materials
Negative control siRNA (NC siRNA) and pooled human HMGI-C siRNA (a pool of 3 different siRNA duplexes sequences including siRNA duplex A (sc-37994A) Sense: GCACUUUCAAUCUCAAUCUtt and Antisense: AGAUUGAGAUUGAAAGUGCtt, siRNA duplex B( sc-37994B) Sense: GUGACCACUUAUUCUGUAUtt and Antisense: AUACAGAAUAAGUGGUCACtt, siRNA duplex C (sc-37994C) Sense: GAGACGAAAUGCUGAUGUAtt and Antisense: UACAUCAGCAUUUCGUCUCtt), goat polyclonal anti HMGI-C anti-body, monoclonal β-actin antibody, siRNA transfection reagent and siRNA transfection medium were purchased from Santa Cruz Biotechnology (California, USA). Monoclonal P53, caspase3, caspase 9, caspase 8 and Bcl2 antibodies were bought from Abcam® (Abcam, Cambridge, MA, USA).
Rabbit anti-goat antibody was purchased from Cytomatin Gene Company (Isfahan, Iran), and rabbit anti-mouse antibody was purchased from Razi Institute. QRT-PCR master mix was purchased from Takara Bio Inc. (Shiga, Japan) and miRNA expression was measured by miRCURY LNA microRNA Reagents (Exiqon), TUNEL and Annexin-V-FLUOS Staining Kit were bought from Roche (Roche, Mannheim, Germany) and propidium iodide dye was purchased from Sigma Aldrich (USA).
Ethics statement
For the analyzed tissue specimens, all patients gave informed consent to use excess pathological specimens for research purposes. The protocols employed in this study and the use of human tissues was approved by the Ethics Committee of Tabriz University of Medical Sciences by the number of 92/74. The manuscript was accompanied by a statement that the experiments were undertaken with the understanding and written consent of each subject and according to the above mentioned principles.
Human tissue specimens and cell line
Fifteen pathologically diagnosed metastatic breast cancer samples and marginal normal tissue samples were acquired from patients with breast cancer, including 4 with distant metastasis. Breast cancer patient tissues were immediately frozen in liquid nitrogen and were stored at −80°C until further use. Cancer tissue samples were obtained from the Emam Reza Hospital affiliated with Tabriz University of Medical Sciences. Human breast cancer cells (MDA-MB-468) and human normal fibroblast cells (HFF-1) were obtained from the Pasteur Institute (Tehran, Iran). Cells were maintained in RPMI 1640 with 10% FBS (GIBCO, Carlsbad, CA, USA) and were cultured at 37°C with 5% CO2. The cells were sub-cultured 24–48 h later with an initial concentration of 4×104 cells/ml and used in the logarithmic phase in all experiments.
In vitro gene silencing
Just before transfection, the cells were cultivated in RPMI-1640 medium free of serum and antibiotics; 2×105 cells were seeded in 6-well cell culture plates and used for each transfection. siRNA transfection (at a final concentration of 80 pmol in all experiments) was performed using siRNA transfection reagent (Santa Cruz Biotechnology, USA) according to the manufacturer's recommendations. Briefly, siRNAs and the siRNA transfection reagent were diluted in siRNA transfection medium (Santa Cruz Biotechnology, USA) separately and incubated for 10 min at room temperature. The diluted solutions were then mixed and incubated for 15–30 min at room temperature. Subsequently, the mixtures were added to each well containing cells and transfection medium. Moreover, the cells treated with only the transfection reagent were considered a blank control. The cell culture plates were then incubated for 5–7 h at 37°C in a CO2 incubator. Following on, RPMI-1640 medium containing FBS (final FBS concentration of 20%) was added, with cells being incubated under the above mentioned conditions.
Quantitative reverse transcriptase polymerase chain reaction analysis (QRTPCR)
Total cellular RNA was isolated by AccuZol reagent (Bioneer, Daedeok-gu, Daejeon, Korea). cDNA (cDNA) was synthesized from 1 μg of total RNA by use of M-MLV reverse transcriptase (Promega, Madison, WI, USA) and random hexamer primer according to Thermo Fisher Scientific manufacturer's instructions. qRT-PCR was performed in the Rotor-Gene 6000 system (Corbett Life Science, Mortlake, NSW, Australia) using SYBR Premix Ex Taq (Takara Bio, Otsu, Shiga, Japan). The reaction system of PCR was: 5 μl of SYBR green reagent, 0.2 μM of each primer, 1 μl of cDNA template and 6 μl of nuclease-free distilled water.
The primer sequences were as follows in .
Table 1. Primer sequences.
The evaluation of HMCI-C, caspase 3, 8, 9 and Bcl2 were performed by initial denaturation step at 94°C for 10 min, followed by 40 cycles at 94°C for 10 sec, 59°C for 30 sec and 72°C for 20 sec, using β-actin as the reference gene. The evaluation of miRNA expression was carried out by initial denaturation step at 95°C for 10 min, followed by 45 cycles at 95°C for 10 sec and 60°C for 60 sec, and miR-103 was used as internal control.Citation41 Relative, mRNAs expression was measured with the 2 −(ΔΔCt) method (Livak and Schmittgen, 2001).
Protein expression assay
Following treatment, the cells were washed twice with cold PBS and lysed on ice in radio immunoprecipitation assay (RIPA) buffer (1% SDS, 1% Triton X-100, 1 mM EDTA, pH 8, 50 mM Tris-HCl, pH 7.4 and 150 mM NaCl) containing protease inhibitor cocktail (Roche Diagnostics GmbH) for 30 min on ice. Suspensions were centrifuged at 14,000 rpm for 10 min at 4°C and cellular debris was discarded. Protein concentrations were quantified using NanoDrop (Thermo Scientific, Wilmington, USA). Fifty micrograms of each protein sample were separated on 12.5% SDS-polyacrylamide gel electrophoresis, transferred to poly vinylidine difluoride membranes (Roche Diagnostics GmbH) and then blocked with 0.5% Tween-20 in PBS/Tween-20 (0.05%, v/v) overnight at 4°C. Following on, the membranes were probed in 1 h at room temperature with polyclonal primary antibodies against HMGI-C (1:2000) and monoclonal antibodies against β-actin (1:5000), p53, caspase 3, 8, 9 and Bcl2 (1:1000) and diluted with 3% BSA in PBS. After four 10 min washes with a buffer containing PBS and 0.05% Tween-20, membranes were incubated with appropriate horse radish peroxidase-linked rabbit anti-goat secondary antibody (1:5000) and rabbit anti-mouse antibody (Razi institute, Tehran, Iran) diluted in PBS and 0.05% Tween-20 for 1 h at room temperature. Subsequently, the membranes were washed and protein bands visualized using enhanced BM chemiluminescence blotting substrate POD (Roche Diagnostics GmbH, Mannheim, Germany) and autoradiography films (Estman Kodak, Rochester, NY, USA).
Cell proliferation assay
The effect of HMGI-C siRNA on the breast cancer cells was determined using 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide (MTT) assay. The experiment was subdivided into 5 groups: non-transfected siRNA (control), negative control siRNA (NC siRNA), pure HMGI-C siRNA (without transfection reagent), transfection reagent (cells treated just by transfection reagent), 40 pmol, 60 pmol and 80 pmol of HMGI-C siRNA. Briefly, cells were cultured at a density of 15×103 cells/well in 96-well cell culture plates and then transfected with siRNAs. After 48 h of incubation, the cytotoxicity of the treatments was assessed using the MTT cell proliferation kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer's protocol. The amount of formazan dye was determined by quantifying its observance (A) at 570 nm (with a reference wavelength of 650 nm) using a microplate reader (Awareness Technology, Palm City, FL, USA). The HMGI-C rate (SR) was measured from the following equation: SR (%) = (A Treatment /A Control) ×100%. The concentration that produced 50% cytotoxicity (IC50) was determined using GraphPad Prism 6.01 software (GraphPad Software Inc., San Diego, CA, USA).
Apoptosis assay
TUNEL assay
The MDA-MB-468 breast adenocarcinoma cells were cultivated at a density of 15×103 cells/well in 96-well plates and then divided into 3 groups: untreated group (control), NC siRNA and 80 pmol HMGI-C specific siRNA, as described in the MTT assay section. After 48 h of incubation, we performed the TUNEL assay; first cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) solution in PBS for 1 h at room temperature, treated with 0.3% H2O2-methanol solution, and then permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate solution for 2 min on ice. The TUNEL assay (Roche Molecular Biochemicals) was carried out following the manufacturer's instruction.
Annexin V/PI assay
Annexin-V/PI assay was used to identify the viable, apoptotic or necrotic cells. The MDA-MB-468 breast adenocarcinoma cells were cultivated at a density of 2×106 cells/well in 6-well plates in 3 groups: control, NC siRNA and 80 pmol HMGI-C specific siRNA. After 48 h of incubation, Annexin-V/PI assay was performed after 48 h post-treatment, cells were washed twice with PBS and suspended in 100 µl of the supplied binding buffer containing Annexin-V-FITC and PI. After incubation in the dark at room temperature for 15 min, cells were immediately analyzed by flow cytometer.
Cell cycle assay
The MDA-MB-468 cells were cultivated at a density of 2×106 cells/well in 6-well plates in 3 groups: control, NC siRNA and 80 pmol HMGI-C specific siRNA. Cells were harvested 48 h after the transfection, fixed with 50% ethanol, treated with 5 mg/ml RNase A (Bioneer, Daedeok-gu, Daejeon, Korea), stained with 50 µg/ml propidium iodide, and analyzed by flow cytometry for DNA synthesis and cell cycle status (Partec, Germany, with FlowJo software).
Statistical analysis
All data in this study was presented as mean ± standard deviation (SD). Statistical significance of differences between groups was explored by using student t-test and analysis of variance (ANOVA) using GraphPad Prism software. The value of P less than 0.05 was considered significant.
Figure 7. schematic model of the HMGI-C network in Breast adenocarcinoma.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dariush Shaneh Bandi and Lila Mohammadnejad for excellent technical support.
References
- DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin 2014; 64:52-62; PMID:24114568
- Whelan J. First clinical data on RNAi. Drug Discov Today 2005; 10:1014-5; PMID:16055013; http://dx.doi.org/10.1016/S1359-6446(05)03547-6
- Fire A. Gene silencing by double-stranded RNA. Cell Death Differ 2007; 14:1998-2012; PMID:18007671; http://dx.doi.org/10.1038/sj.cdd.4402253
- Kachalaki S, Baradaran B, Majidi J, Yousefi M, Shanehbandi D, Mohammadinejad S, Mansoori B. Reversal of chemoresistance with small interference RNA (siRNA) in etoposide resistant acute myeloid leukemia cells (HL-60). Biomed Pharmacother 2015; 75:100-4; PMID:26463638; http://dx.doi.org/10.1016/j.biopha.2015.08.032
- Montazami N, Kheirandish M, Majidi J, Yousefi M, Yousefi B, Mohamadnejad L, Shanebandi D, Estiar M, Khaze V, Mansoori B. siRNA-mediated silencing of MDR1 reverses the resistance to oxaliplatin in SW480/OxR colon cancer cells. Cell Mol Biol (Noisy-le-Grand, France) 2015; 61:98; PMID:26025411
- Mansoori B, Shotorbani SS, Baradaran B. RNA Interference and its Role in Cancer Therapy. 2014.
- Mansoori B, Mohammadi A, Shir Jang S, Baradaran B. Mechanisms of immune system activation in mammalians by small interfering RNA (siRNA). Artif Cells Nanomed Biotechnol 2015:1-8; PMID:26497011; http://dx.doi.org/10.3109/21691401.2015.1102738
- Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006; 126:503-14; PMID:16901784; http://dx.doi.org/10.1016/j.cell.2006.05.052
- Reeves R. Molecular biology of HMGA proteins: hubs of nuclear function. Gene 2001; 277:63-81; PMID:11602345; http://dx.doi.org/10.1016/S0378-1119(01)00689-8
- Natarajan S, Hombach-Klonisch S, Dröge P, Klonisch T. HMGA2 inhibits apoptosis through interaction with ATR-CHK1 signaling complex in human cancer cells. Neoplasia 2013; 15:263-IN13; PMID:23479505; http://dx.doi.org/10.1593/neo.121988
- Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996; 13:2439-46; PMID:8957086
- Hirning‐Folz U, Wilda M, Rippe V, Bullerdiek J, Hameister H. The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer 1998; 23:350-7; http://dx.doi.org/10.1002/(SICI)1098-2264(199812)23:4%3c350::AID-GCC10%3e3.0.CO;2-E
- Gattas GJ, Quade BJ, Nowak RA, Morton CC. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 1999; 25:316-22; http://dx.doi.org/10.1002/(SICI)1098-2264(199908)25:4%3c316::AID-GCC2%3e3.0.CO;2-0
- Rogalla P, Drechsler K, Frey G, Hennig Y, Helmke B, Bonk U, Bullerdiek J. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am J Pathol 1996; 149:775; PMID:8780382
- Meyer B, Loeschke S, Schultze A, Weigel T, Sandkamp M, Goldmann T, Vollmer E, Bullerdiek J. HMGA2 overexpression in non-small cell lung cancer. Mol Carcinog 2007; 46:503-11; PMID:17477356; http://dx.doi.org/10.1002/mc.20235
- Abe N, Watanabe T, Suzuki Y, Matsumoto N, Masaki T, Mori T, Sugiyama M, Chiappetta G, Fusco A, Atomi Y. An increased high-mobility group A2 expression level is associated with malignant phenotype in pancreatic exocrine tissue. Br J Cancer 2003; 89:2104-9; PMID:14647145; http://dx.doi.org/10.1038/sj.bjc.6601391
- Malek A, Bakhidze E, Noske A, Sers C, Aigner A, Schäfer R, Tchernitsa O. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int J Cancer 2008; 123:348-56; PMID:18452175; http://dx.doi.org/10.1002/ijc.23491
- Guang-meng X, Hai-na Z, Xiao-feng T, Mei S, Xue-dong F. Effect of HMGA2 shRNA on the Cell Proliferation and Invasion of Human Colorectal Cancer SW480 Cells< em>In vitro. Chem Res Chinese Universites 2012; 28:264-8.
- Chau K-Y, Manfioletti G, Cheung-Chau K-W, Fusco A, Dhomen N, Sowden JC, Sasabe T, Mukai S, Ono SJ. Derepression of HMGA2 gene expression in retinoblastoma is associated with cell proliferation. Mol Med 2003; 9:154; PMID:14571323; http://dx.doi.org/10.2119/2003-00020.Ono
- Miyazawa J, Mitoro A, Kawashiri S, Chada KK, Imai K. Expression of mesenchyme-specific gene HMGA2 in squamous cell carcinomas of the oral cavity. Cancer Res 2004; 64:2024-9; PMID:15026339; http://dx.doi.org/10.1158/0008-5472.CAN-03-1855
- Andrieux J, Demory JL, Dupriez B, Quief S, Plantier I, Roumier C, Bauters F, Laï JL, Kerckaert JP. Dysregulation and overexpression of HMGA2 in myelofibrosis with myeloid metaplasia. Genes Chromosomes Cancer 2004; 39:82-7; http://dx.doi.org/10.1002/gcc.10297
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 2007; 21:1025-30; PMID:17437991; http://dx.doi.org/10.1101/gad.1540407
- Thuault S, Tan E-J, Peinado H, Cano A, Heldin C-H, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 2008; 283:33437-46; PMID:18832382; http://dx.doi.org/10.1074/jbc.M802016200
- Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol Cancer 2007; 6:5; PMID:17222355; http://dx.doi.org/10.1186/1476-4598-6-5
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007; 315:1576-9; PMID:17322030; http://dx.doi.org/10.1126/science.1137999
- Mansoori B, Mohammadi A, Shirjang S, Baradaran B. Micro-RNAs: The new potential biomarkers in cancer diagnosis, prognosis and cancer therapy. Cell Mol Biol (Noisy-le-Grand, France) 2014; 61:1-10.
- Ma Z-L, Hou P-P, Li Y-L, Wang D-T, Yuan T-W, Wei J-L, Zhao B-T, Lou J-T, Zhao X-T, Jin Y. MicroRNA-34a inhibits the proliferation and promotes the apoptosis of non-small cell lung cancer H1299 cell line by targeting TGFβR2. Tumor Biol 2015; 36:2481-90; PMID:25501507; http://dx.doi.org/10.1007/s13277-014-2861-5
- Li L, Wu J, Sima X, Bai P, Deng W, Deng X, Zhang L, Gao L. Interactions of miR-34b/c and TP-53 polymorphisms on the risk of nasopharyngeal carcinoma. Tumor Biol 2013; 34:1919-23; PMID:23504554; http://dx.doi.org/10.1007/s13277-013-0736-9
- Cao W, Fan R, Wang L, Cheng S, Li H, Jiang J, Geng M, Jin Y, Wu Y. Expression and regulatory function of miRNA-34a in targeting survivin in gastric cancer cells. Tumor Biol 2013; 34:963-71; PMID:23264087; http://dx.doi.org/10.1007/s13277-012-0632-8
- Karami H, Baradaran B, Esfahani A, Sakhinia M, Sakhinia E. Therapeutic Effects of Myeloid Cell Leukemia-1 siRNA on Human Acute Myeloid Leukemia Cells. Adv Pharm Bull 2014; 4:243; PMID:24754007
- Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist Updat 2002; 5:65-72; PMID:12135582; http://dx.doi.org/10.1016/S1368-7646(02)00049-3
- Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res 2006; 12:11-9; PMID:16397018; http://dx.doi.org/10.1158/1078-0432.CCR-04-1752
- HOECKER P, FISCHER MB, JAKESZ R, ZEILLINGER R. HMGA2 is associated with invasiveness but not a suitable marker for the detection of circulating tumor cells in breast cancer. Oncol Rep 2005; 14:737-41; PMID:16077985
- Motoyama K, Inoue H, Nakamura Y, Uetake H, Sugihara K, Mori M. Clinical significance of high mobility group A2 in human gastric cancer and its relationship to let-7 microRNA family. Clin Cancer Res 2008; 14:2334-40; PMID:18413822; http://dx.doi.org/10.1158/1078-0432.CCR-07-4667
- Sarhadi V, Wikman H, Salmenkivi K, Kuosma E, Sioris T, Salo J, Karjalainen A, Knuutila S, Anttila S. Increased expression of high mobility group A proteins in lung cancer. J Pathol 2006; 209:206-12; PMID:16521118; http://dx.doi.org/10.1002/path.1960
- Wang Q, Gong Y, Lü Y, Fei L, Liu H, Diao Y, Xu R. [Selection and anti-cancer effects of siRNAs targeting HMGA2 gene]. Yao Xue Xue Bao 2011; 46:1444-50; PMID:22375416
- Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1, and p53: the feedback loop. Cell Cycle 2009; 8:712-5; PMID:19221490; http://dx.doi.org/10.4161/cc.8.5.7753
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17:193-9; PMID:19461653; http://dx.doi.org/10.1038/cdd.2009.56
- Heldin C-H, Landström M, Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 2009; 21:166-76; PMID:19237272; http://dx.doi.org/10.1016/j.ceb.2009.01.021
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2009; 17:193-9; PMID:19461653; http://dx.doi.org/10.1038/cdd.2009.56
- Song J, Bai Z, Han W, Zhang J, Meng H, Bi J, Ma X, Han S, Zhang Z. Identification of suitable reference genes for qPCR analysis of serum microRNA in gastric cancer patients. Dig Dis Sci 2012; 57:897-904; PMID:22198701; http://dx.doi.org/10.1007/s10620-011-1981-7