
Keywords:
Tumor suppressor p53 plays an essential role in preventing cancer development. In healthy cells p53 levels are kept low by degradation via the ubiquitin ligase Mdm2, whose gene expression is, in turn, activated by p53. The TP53 gene is mutated in about 50% of human cancers. In addition, oncogenic viruses have developed strategies to inactivate p53 functions in both infected and transformed cells. Viral oncoproteins such as Polyomavirus SV40 Large T antigen (LTag), Adenovirus E1B, Epstein-Barr virus EBNA-5 bind to p53 in a manner that increases the cellular levels of p53 while inhibiting its transcriptional functions.Citation1 By contrast, the E6 oncoprotein from ‘high-risk’ mucosal (hrm) Human Papillomaviruses (HPV), responsible for cervical carcinomas and a growing number of head-and-neck cancers, forms a ternary complex with the cellular E3 ubiquitin ligase E6AP and p53. This complex leads to ubiquitin-mediated degradation of p53 and subsequent decrease of its cellular levels.Citation2
Despite the early discovery in the nineties of HPV E6-mediated degradation of p53, no structural information on this process existed until recently, mainly because of the poor stability of recombinant HPV E6 samples. However, it was well-established that most mammalian Papillomavirus (PV) E6 proteins can recruit target proteins by recognition of acidic-leucine (L)-rich-LxxLL sequence motifs. Such motifs are found within E6AP as well as in other cellular E6 targets including interferon regulatory factor-3 (IRF-3), the notch co-activator MAML1, and the focal adhesion protein paxillin. By contrast neither E6 nor E6AP are separately able to interact with p53. A recent key observation has been that a peptide comprising the LxxLL motif of E6AP is sufficient to render E6 liable to interact with p53.Citation3 This suggested that prior formation of an HPV E6/E6AP heterodimer was required for E6 binding to p53.
Biophysical studies on E6 self-association combined with structural data on the isolated E6 zinc-binding domains (E6N and E6C) from HPV16 (the most prevalent and best studied hrm-HPV) pointed to strategies to enhance E6 solubility. In this way we created a soluble construct of HPV16 E6 comprising 5 point mutations, which did not alter E6 structure.Citation4 These findings, together with strategies based on fusion carrier proteins, opened the way to structural studies of complexes of HPV E6.
We first solved the x-ray structure of HPV16 E6 bound to a peptide comprising the LxxLL motif of E6AP (called e6ap from hereon).Citation5 In the same study we also reported the crystal structure of the E6 oncoprotein from Bovine Papillomavirus 1 (BPV1) bound to the LxxLL sequence from paxillin. In both complexes the LxxLL motif adopts a helical conformation and binds a hydrophobic-basic pocket, which is contributed by residues from the E6N and E6C domains, and from the interdomain helical linker. Sequence alignments coupled to mutagenesis and binding assays clearly indicate that the LxxLL pocket is conserved among E6 proteins issued from mammalian Papillomaviruses.
More recently, we were able to solve the crystal structure of a minimal ternary complex, comprising HPV16 E6, e6ap and the core (or DNA binding) domain of p53 (called p53core from hereon)Citation6 (, top and middle). In this complex structure p53core binds to a cleft formed by the E6N and E6C domains that are tethered to opposite sides of the helical e6ap peptide. By contrast, despite being absolutely required for E6 binding to p53, e6ap does not contribute significant intermolecular contacts to p53core. Notably, the structure of the E6/e6ap subunit is unaffected by p53 binding. Altogether, these observations indicate that the e6ap peptide acts as a conformational switch, which structures the p53-binding cleft on E6. This suggests that, besides the LxxLL motif of E6AP, other peptide sequences targeting the LxxLL pocket may also promote p53 recruitment. Indeed this is the case of a previously in vitro selected peptide, which induces p53 binding to HPV16 E6.Citation7 In the study describing the E6/e6ap/p53core complex we were also able to identify sequence features that drive p53 degradation within the hrm-HPV group. In particular, residues within the α2 helix of E6 (residues 40–46), which establish crucial contacts to p53, are conserved in hrm-HPV E6 sequences but not in low-risk mucosal HPV E6 proteins, which do not degrade p53.
Figure 1. (Top) Ribbon representation of the HPV16 E6/e6ap/p53core ternary complex. Gold: HPV16 E6; green: e6ap; purple: p53core. (Middle) Surface representation of HPV16 E6 colored for residues at the interface with e6ap (green), p53 (purple) and e6ap and p53 (gray). E6N and E6C: N- and C-terminal zinc-binding domains; HL: helix linker. (Bottom) Surface representation of p53core colored for residues at the interface with HPV E6 (gold), DNA (cyan) and SV40 LTag (pink).
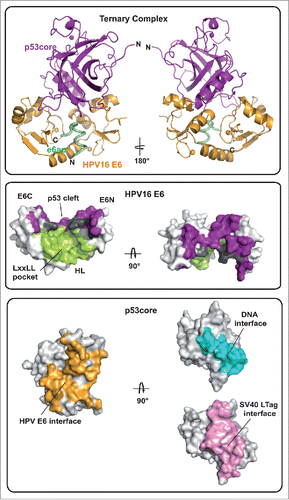
The only other viral oncoprotein for which structural data are available on the p53core targeting mechanism is SV40 LTag (PDB:2H1L). Whereas SV40 LTag binds to the DNA binding interface of p53, thereby leading to inhibition of p53 transactivation functions, HPV E6 targets a completely distinct p53 interface (, bottom). Analysis of other complex structures of p53core shows that the E6-binding surface of p53 is available for interaction not only in ‘free’ p53 tetramers but also in p53 molecules bound to DNA or other cellular proteins. Hence, this ability to target several species of cellular p53 may render E6 a potent inactivator of p53 functions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Levine AJ. Virology 2009; 384:285-93; PMID:19081592; http://dx.doi.org/10.1016/j.virol.2008.09.034
- Werness BA, et al. Science 1990; 248:76-79; PMID:2157286;
- Ansari T, et al. J Virol 2012; 20:11386-91; PMID:22896608;
- Zanier K, et al. Structure 2012; 20:604-17; PMID:22483108; http://dx.doi.org/10.1016/j.str.2012.02.001
- Zanier K, et al. Science 2013; 339:694-8; PMID:23393263; http://dx.doi.org/10.1126/science.1229934
- Martinez-Zapien D, et al. Nature 2016; 529:541-5; PMID:26789255; http://dx.doi.org/10.1038/nature16481
- Stutz C, et al. PLoS One 2015; 10:e0132339; PMID:26151636; http://dx.doi.org/10.1371/journal.pone.0132339