
ABSTRACT
ADP-ribosylation is involved in a variety of biological processes, many of which are chromatin-dependent and linked to important functions during the cell cycle. However, any study on ADP-ribosylation and the cell cycle faces the problem that synchronization with chemical agents or by serum starvation and subsequent growth factor addition already activates ADP-ribosylation by itself. Here, we investigated the functional contribution of ARTD1 in cell cycle re-entry and G1/S cell cycle progression using T24 urinary bladder carcinoma cells, which synchronously re-enter the cell cycle after splitting without any additional stimuli. In synchronized cells, ARTD1 knockdown, but not inhibition of its enzymatic activity, caused specific down-regulation of cyclin E during cell cycle re-entry and G1/S progression through alterations of the chromatin composition and histone acetylation, but not of other E2F-1 target genes. Although Cdk2 formed a functional complex with the residual cyclin E, p27KipCitation1 protein levels increased in G1 upon ARTD1 knockdown most likely due to inappropriate cyclin E-Cdk2-induced phosphorylation-dependent degradation, leading to decelerated G1/S progression. These results provide evidence that ARTD1 regulates cell cycle re-entry and G1/S progression via cyclin E expression and p27KipCitation1 stability independently of its enzymatic activity, uncovering a novel cell cycle regulatory mechanism.
Introduction
The G0 phase of the cell cycle is a transcriptionally silent stage cells enter upon growth inhibition.Citation1 When the conditions become favorable for cell growth again, cells re-enter the G1 phase and progress through the cell cycle. Among the molecular factors involved in cell cycle re-entry are transcription factors of the E2F family (e.g., E2F1, E2F4), cyclins, cyclin-dependent kinases (e.g., Cdk2, Cdk3, Cdk4, Cdk6),Citation2 the retinoblastoma protein (pRb),Citation3 and microRNAs.Citation4 pRb is one of the key players of the early cell cycle phases, whose inactivation by phosphorylation seems to be sufficient for cell cycle re-entry.Citation5 For example, pRb is hyperphosphorylated in asynchronous human T24 bladder carcinoma cells and thus favors cell cycle progression.Citation6 The most prominent factors regulated by pRb are histone deacetylases, chromatin remodeling complexes and the transcription factors E2F.Citation7 E2F proteins are a family of transcription factors of which E2F-1 to 3 are activators and E2F-4 to 8 are repressors. E2F target genes include cyclin E, A, D1, Cdc2, Cdc25A, DNA polymerase, Cdc6 and minichromosome maintenance (MCM).Citation8 Cyclin E, in complex with Cdk2, is the main driver of G1/S phase progression. Cdk inhibitors such as the CIP/KIP family proteins p21 and p27 are negative regulators that bind to cyclin-Cdk complexes and inhibit their enzymatic activity.Citation9 p27 has previously been identified as specific cyclin E/Cdk2 phosphorylation target, and p27 phosphorylation to result in the elimination of p27 from the cell, allowing the transit from G1 to the S phase.Citation10
ADP-ribosylation is a post-translational modification (PTM) catalyzed by extra- and intracellular ADP-ribosyltransferases (ARTCs and ARTDs, respectively), whereby the ADP-ribose moiety of NAD+ is transferred to specific amino acid residues, resulting in mono- or poly-ADP-ribosylation of target proteins. The human ARTD family consists of 18 members, of which ARTD1 (formerly PARP1) is the best-studied member. ARTD1 is a nuclear protein that poly-ADP-ribosylates both itself and target proteins. ARTD1 is the most active cellular ADP-ribosyltransferase and contributes to 90-95 % of total poly-ADP-ribose (PAR) synthesis.Citation11
ADP-ribosylation is involved in a variety of biological processes, many of which are chromatin-dependent and linked to important functions during the cell cycle. ARTD1 localizes to and modifies centromeric proteins,Citation12-14 and regulates the mitotic chromosomal protein kinase aurora B by PARylation.Citation15 Importantly, depletion of ARTD1 or inhibition of ADP-ribosylation has been shown to lead to a cell cycle arrest in the prophase or prolongation of the G2-M phase transition, suggesting that it is required for chromosomal functions during mitosis.Citation16-20 Recently, it has also been shown that activation of ARTD1 via Cdk2-dependent phosphorylation and consecutive modification and displacement of histone H1 is involved in progestin gene regulation.Citation21 This is essential for the effect of progestins on cell cycle progression in breast cancer cells. There are also various reports that have implicated functions of ARTD1 in early cell cycle phases. For example, ARTD1 and the C-terminal binding protein (CtBP) form a co-repressor complex for p21 repression, and ARTD1 activity is necessary for p21 activation after genotoxic stress.Citation22 Simbulan-Rosenthal et al. have shown that ARTD1 upregulates the promoter activity of E2F-1 during the early S phase, and that ARDT1 interacts with E2F-1 both in vitro and in immortalized fibroblasts synchronized by serum deprivation or aphidicolin treatment.Citation23,24 Another study has detected PAR formation during G0-G1 transition after mitogen stimulation and has thus implicated ARTD1 in growth factor signaling and the induction of immediate-early genes.Citation25 More generally, ADP-ribosylation is involved in spindle formation, the assembly of spindle poles,Citation26,27 heterochromatin formation,Citation28 and the maintenance of an ATR and Chk1-dependent S-phase checkpoint.Citation29
Any study on ADP-ribosylation and the cell cycle faces the problem that synchronization with chemical agents or by serum starvation and subsequent growth factor addition activates ADP-ribosylation itself.Citation30 In the study described here, we used T24 urinary bladder carcinoma cells, which represent a well-characterized, exemplary bladder cancer cell line.Citation31,32 Importantly, T24 cells synchronously re-enter the cell cycle after splitting without any additional stimulation and are therefore an ideal model to study cell cycle re-entry and G1/S regulation.
Here, ARTD1 was down-regulated in T24 cells using an siRNA approach and its effect were studied on cell cycle re-entry and G1/S progression. Knockdown of ARTD1 in T24 cells caused downregulation of E2F-1-dependent cyclin E expression via increased H1 recruitment, reduced H4 acetylation, as well as a lower H2A.Z content at the cyclin E promoter. Interestingly, the expression of other E2F-1 target genes was not affected or was even enhanced. Knockdown of ARTD1 also led to increased p27 levels, a known regulatory mechanism that consequently led to a decelerated cell cycle entry and G1/S progression.Citation10 Pharmacological inhibition of ADP-ribosylation did not affect cell-cycle re-entry, but instead led to an increase in the cell population in S-phase and a reduction in G2/M-phase cells. These molecular analyses thus confirm the regulation of cyclin E as a novel mechanism of cell cycle re-entry and G1/S progression regulation by ARTD1, independent of its enzymatic activity.
Results
ARTD1 is involved in cell cycle re-entry and G1/S progression of T24 cells
To elucidate the role of ARTD1 protein and its enzymatic activity on cell cycle re-entry and G1/S progression, the urinary bladder carcinoma cell line T24 was used. T24 cells are characterized by arresting in the G0 phase of the cell cycle upon reaching confluence, and synchronously re-entering the cell cycle after splitting for one cell cycle, without additional stimulation.Citation33,34 These cells are thus an ideal model, because synchronization does not require chemical agents or stimuli that may activate ADP-ribosylation per se. Confluent T24 cells (i.e., contact-inhibited monolayers) are characterized by strong, cell-cycle dependent retinoblastoma (Rb) phosphorylation,Citation6 as compared to confluent U2OS cells (). Any cell cycle effects in T24 cells are thus down-stream and independent of Rb phosphorylation. To assess the role of ARTD1 in cell cycle re-entry and G1/S progression, ARTD1 in T24 cells was knocked down in G0, which were subsequently synchronously released into the cell cycle by splitting the cells (Fig. S1A). While the expression of ARTD1 remained knocked down during the whole analysis ( and S1B), total Rb protein levels were not altered (Fig. S1C).
Figure 1. ARTD1 knockdown leads to cell cycle delay in the T24 hyperphosphorylated cell line. A) Western blot analysis with extracts of confluent T24 and U2OS cells to assess the levels of Rb phosphorylation. B) Western blot confirmation of ARTD1 depletion upon siRNA treatment of T24 cells. C) Flow cytometry analysis of siMock and siARTD1-treated, synchronized T24 cells at different time points after release from G0 phase and asynchronous T24 cells. D) Quantification of cell cycle analysis after knockdown of ARTD1 (A1) shown in C. Data represent mean ± SD of n = 4 independent experiments and was analyzed by 2-way-ANOVA followed by a Bonferroni post-test for the G1 and S phase. *P < 0.05; ***P < 0.001: A1 statistically different to corresponding siMock (M)-treated cells at the same time point. E) Flow cytometry analysis for shMock and shARTD1 transduced T24 cells shortly after viral transduction. F) Immunofluorescence microscopy of PAR formation of untreated or H2O2-treated (10 min, 1 mM) samples, pre-treated with olaparib (1 µM) or veliparib (1 µM). G) Inhibition of the enzymatic activity leads to an S phase arrest. Overlay of flow cytometry samples of T24 cells: untreated (non-transduced), olaparib (24 h, 1 µM) and veliparib (24 h, 1 µM) treated samples (24 h time point).
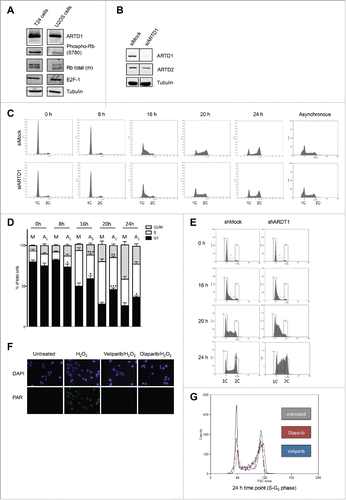
Flow-cytometry analysis of siMock and siARTD1-treated T24 cells indicated a delayed cell cycle re-entry and progression for ARTD1 knocked-down cells, which suggests a decelerated G1/S transition (). Thus, between 16 and 24 h after re-seeding, significantly more siARTD1-treated cells were in the G1 phase compared to siMock-treated cultures, and significantly less cells were in the S phase. This finding was confirmed by reduced bromodeoxyuridine (BrdU) incorporation in ARTD1 knocked-down cells during the first 16 h (Fig. S1D). No effect of ARTD1 knock-down on cell cycle distribution was observed when asynchronous T24 cells were used, suggesting that ARTD1 mainly regulates cell cycle re-entry and G1/S progression and that the described delay is only observed with synchronized cells (). Stably knocking down ARTD1 in T24 by shRNA caused an even stronger delay in G1/S transition (). Treatment of G0 T24 cells with either the PARP inhibitor olaparib or veliparib at concentrations that completely abolish H2O2-induced PAR formation in these cells () and subsequent release into the cell cycle did not affect cell-cycle re-entry, but rather led to an increase in S phase cells and a reduction in G2/M phase cells (). These results indicate that ADP-ribosylation is not required for cell cycle re-entry and G1 progression, but rather is required for progression through the S phase. Altogether, these results suggest that ARTD1 plays an important role in cell cycle re-entry and G1/S progression of arrested T24 urinary bladder carcinoma cells, but independent of its enzymatic activity.
ARTD1 down-regulation reduces cyclin E expression
As ARTD1 has been implicated in DNA repair, siARTD1 treatment of T24 cells may induce DNA damage and thereby affect cell cycle progression. Western blot analysis of p53 stability, replication protein A (RPA), and Chk-1 phosphorylation from siMock and siARTD1-treated cells at different time points after release from confluency indicated that knock-down of ARTD1 in T24 cells did not activate the DNA damage response ( and Fig. S2A). When analyzing γH2AX levels by Western blot and immunofluorescence microscopy, we observed enhanced γH2AX formation when ARTD1 levels were knocked down by siRNA in confluent T24 cells, which was markedly reduced when cells were released into the cell cycle, and only reappeared when cells reached the S phase ( and Fig. S2B). Interestingly, when analyzing confluent siARTD1-treated T24 cells by comet assay, no DNA breaks were observed (Fig. S2C). Thus, although the exact cause of elevated γH2AX levels remains to be determined, they did not activate the DNA damage response.
Figure 2. Cyclin E protein levels are reduced upon ARTD1 depletion. A-B) Western blot analysis of p53 (A) and γH2AX (B) after cell cycle re-initiation of synchronized siMock (M) or siARTD1 (A1) treated T24 cells at different time points. As positive control (T24*/U2OS), cells treated with etoposide (10 µM, 16 h) were used. C) qPCR analysis of cyclin A, B and D1 in siMock and siARTD1-treated cells. (n = 2) D) qPCR analysis of cyclin E in siMock and siARTD-treated T24 cells (n = 4, t-test). E) Western blot analysis of cyclin E levels in siMock and siARTD1 treated cells. F) qPCR analysis of cyclin E expression in cells treated with the PARP inhibitor olaparib 1 μM (n = 2). (G) qPCR analysis of asynchronous siMock- and siARTD1-treated T24 cells.
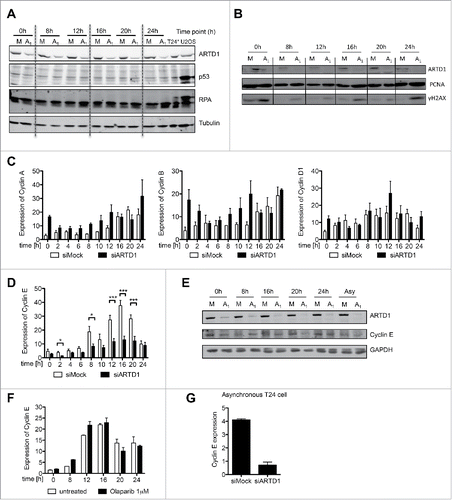
To characterize and mechanistically understand the function of ARTD1 in cell cycle progression and chromatin regulation of T24 cells, the gene expression of cyclin A, B, D and E was analyzed. Interestingly, while cyclin A, B and D transcript and protein levels were unchanged or slightly increased upon knockdown of ARTD1 (, Fig. S2D), mRNA and protein levels of cyclin E were significantly lower at all time-points measured ( and E), suggesting that ARTD1 regulates cyclin E expression. Stably knocking down ARTD1 in T24 by shRNA reduced the expression of cyclin E comparably to transient knockdown with siRNA (Fig. S2E). In contrast, treatment of T24 cells with olaparib did not affect cyclin E expression at any time during the cell cycle (), again confirming that mitigation of ADP-ribosylation is not involved in the reduced cyclin E expression in ARTD1 knocked-down cells. Interestingly, although cyclin E was transcriptionally downregulated by siARTD1 also in asynchronous cells (), cyclin E protein levels were not reduced to the same extent, providing further evidence that synchronization of cells is required for the observed reduction in cell cycle-re-entry and subsequent G1/S progression (, last 2 lanes). These findings thus reveal that ARTD1 depletion specifically reduces cyclin E expression and protein levels in synchronized T24 cells released from arrest.
The function of the transcription factor E2F-1 is not impaired upon ARTD1 knockdown
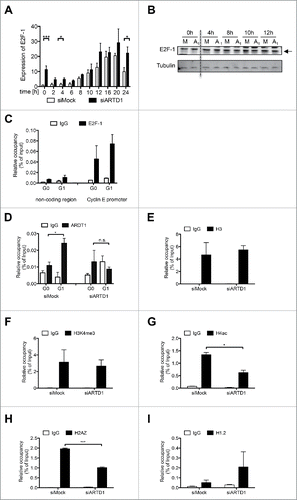
To elucidate the mechanisms by which ARTD1 regulates expression of cyclin E, we analyzed the expression and function of the transcription factor E2F-1, the major regulator of cyclin E expression.Citation35 While E2F-1 transcript levels were significantly elevated in siARTD1-treated cells compared to siMock-treated cells at several time-points (0, 4, and 24 h) (), these changes were not observed at the protein level (). Furthermore, we analyzed binding of E2F-1 to the cyclin E promoter in T24 cells and found that it was present in both G0 and G1 (). Since reduced cyclin E expression could not be attributed to either altered E2F-1 expression or its recruitment, we analyzed the expression of other E2F-1 target genes. Interestingly, expression of the E2F target gene c-myc as well as miR-15 and miR-16 upon siARTD1 treatment were up-regulated rather than repressed (Fig. S3A, S3B, S3C), suggesting that the observed reduction of cyclin E by ARTD1 is gene-specific.
Figure 3. ARTD1 is recruited to the cyclin E promoter and keeps the chromatin in an open conformation. A-B) qPCR analysis (n = 4, t-test) (A) and Western blot analysis (B) of E2F-1 in synchronized siMock and siARTD1-treated T24 cells. C) ChIP analysis of E2F-1 antibody binding to the cyclin E promoter in T24 cells during cell cycle progression. D-I) ChIP analysis of the cyclin E promoter in siMock and siARDT1-treated T24 cells. ARTD1 binding (D), and H3 occupancy (E), H3K4me3 (F), H4 acetylation (G), H2AZ occupancy (I), and H1.2 occupancy (J) during the G1 phase (10 h time point) were analyzed.
ARTD1 renders the cyclin E promoter permissive for transcription
To elucidate how ARTD1 knock-down reduces cyclin E expression, repressory and activatory marks at the cyclin E promoter were analyzed. In agreement with the findings so far, the recruitment of ARTD1 to the cyclin E promoter was significantly increased upon entry of siMock-treated T24 cells into the G1 phase (). In siARTD1 cells, no recruitment was detected, suggesting that ARTD1 might render the cyclin E promoter permissive and therefore transcriptionally active. siARTD1 treatment did not affect histone H3 levels at the cyclin E promoter during G1 (). Interestingly, while H3 lysine 4 trimethylation (H3K4me3), a mark for actively transcribed genes, remained unchanged during G1, histone H4 acetylation (H4ac), another mark of active transcription, was strongly reduced in siARTD1 T24 cells compared to siMock-treated cells (), suggesting that the chromatin at the cyclin E promoter was in a rather condensed state. Furthermore, another mark associated with promoters of actively transcribed genes, the histone H2A variant H2A.ZCitation36 was significantly reduced upon the depletion of ARTD1 by siRNA compared to siMock-treated cells in G1 (). In contrast, the linker histone variant H1.2 was rather increased at the cyclin E promoter upon siARTD1 treatment in G1 (). In summary, the knock-down of ARTD1 enhances compaction of the cyclin E promoter (loss of H4 acetylation, increase of H1 recruitment) and subsequently represses the transcription of cyclin E (loss of H2A.Z). We therefore conclude that ARTD1 regulates cyclin E expression indirectly via the modification of the chromatin composition of the cyclin E promoter region.
Knockdown of cyclin E phenocopies the cell cycle entry and G1/S progression profile observed by knockdown of ARTD1 in T24 cells
To confirm that ARTD1 regulates cell cycle re-entry mainly through cyclin E expression, cyclin E was knocked down and cell cycle progression analyzed (Fig. S3D). siRNA-mediated downregulation of cyclin E in T24 cells had a similar effect on cell cycle re-entry and G1/S progression as siARTD1 treatment did (cf. ), albeit to a somewhat lesser degree (). This apparent discrepancy is most probably due to the less complete down-regulation of cyclin E by the siCyclin E treatment (Fig. S3D). Importantly, cyclin E downregulation had no effect on ARTD1 levels (Fig. S3D), indicating that in this experiment, cyclin E acted down-stream of ARTD1. Based on these results, we conclude that enhanced cyclin E expression is the main contributor of ARTD1-regulated cell cycle re-entry and G1/S progression in T24 cells.
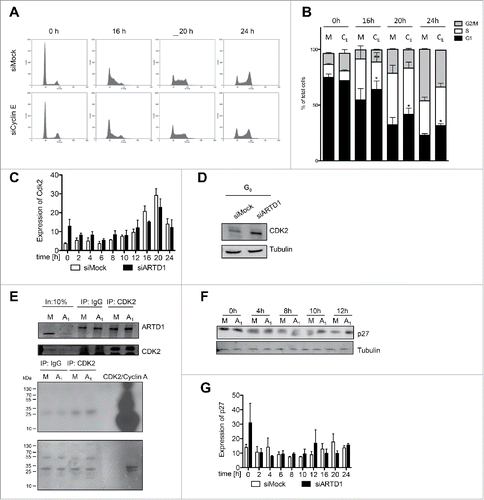
Figure 4. Reduced functional activity of the cyclin E/Cdk2 complex and the consequent increase in p27 levels account for the reduced cell cycle entry in ARTD1-depleted T24 cells. A) FACS analysis of synchronized siMock, siARDT1 and siCyclin E-treated cells during cell cycle progression. B) Quantification of cell cycle analysis after knockdown of cyclin E (CE) shown in C. Data represent mean ± SD of n = 3 independent experiments and was analyzed by 2-way-ANOVA followed by a Bonferroni post-test for the G1 and S phase. *P< 0.05; **P<0.01 CE statistically different to corresponding siMock (M)-treated cells at the same time point. C) qPCR analyses of Cdk2 in siMock and siARTD1-treated samples (n = 4). D) Western blot analysis of Cdk2 in synchronized siMock (M) and siARTD1 (A1)-treated cells. E) Activity assay of Cdk2-cyclin E, immunoprecipitation of Cdk2 at 12 h after re-entry into the cell cycle (upper panel) and radioactive assay with immunoprecipitated Cdk2 or IgG control. Radioactive ATP was used for the assay (lower panel). F) Western blot analysis of p27 protein levels in control and siARTD1 samples (n = 2). G) qPCR analysis of p27KIPin siMock and siARTD1-treated samples.
Knockdown of ARTD1 does not alter Cdk2 levels, but leads to enhanced p27 levels in G1
Next, we aimed to elucidate the functional consequence of cyclin E down-regulation for the reduced cell cycle entry and progression in ARTD1 knocked-down T24 cells. Since cyclin E is known to interact and stimulate Cdk2 activity, we analyzed whether Cdk2 levels were affected by knockdown of ARTD1. While treatment of cells with siARTD1 did not affect expression of Cdk2, protein levels were slightly increased, which cannot account for the observed deceleration in cell cycle progression, however (). In agreement with this result, the in vitro activity of Cdk2 immunoprecipitated from siARTD1-treated cells 12 h after re-entering the cell cycle was not significantly different compared to that from corresponding siMock-treated cells (), suggesting that Cdk2 per se was able to form an active complex with cyclin E in siARTD1-treated cells.
To investigate whether the observed cyclin E/Cdk2-dependent activity is at the functional level comparable in siMock and siARTD1 cells, we analyzed the function of the cyclin E/Cdk2 complex in vivo. Phosphorylation of p27 at T187 is an important activity of the cyclin E/Cdk2 complex.Citation10 Cyclin E/Cdk2-dependent phosphorylation of p27 results in elimination of p27 through protein degradation, which allows cells to transit from G1 to the S phase.Citation37 Increased p27 levels could thus explain the observed delay in G1/S progression (). Indeed, Western blot analysis of cells with reduced ARTD1 levels revealed increased p27 levels, particularly during the G1 phase (), while p27 expression at the corresponding time points was not affected by siARTD1 treatment (). These results suggest that the observed increase in p27 protein levels is not due increased p27 expression, but rather due to post-transcriptional protein stabilization as a result of the reduced functional activity of the cyclin E/Cdk2 complex.
Discussion
T24 bladder carcinoma cells re-enter the cell cycle and divide synchronously after splitting and are therefore an ideal model for studying cell cycle regulation without the need for addition of toxins or stimulatory factors. The use of an siRNA approach to down-regulate ARTD1 in T24 cells and an analysis of cell cycle re-entry and G1/S progression of synchronous cells revealed that ARTD1 alters the chromatin composition, resulting in downregulation of cyclin E expression. The molecular analyses of marks at the promoter sites of cyclin E in siARTD1-treated T24 cells revealed that ARTD1 keeps the chromatin at the cyclin E promoter site permissive for transcriptional activation. Knockdown of cyclin E phenocopied the delayed cell cycle re-entry and G1/S progression observed when ARTD1 was knocked down. These results therefore confirm non-redundant functions in cell cycle re-entry and G1/S progression through differential gene expression regulated by ARTD1.
Our results indicate that ARTD1 is required for the enhanced regulation of cyclin E expression during cell cycle re-entry and G1/S progression. The increased recruitment of ARTD1 to the cyclin E promoter during G0/G1 phase transition suggests that ARTD1 might affect the initiation of cyclin E transcription. Interestingly, this effect was independent of the enzymatic activity of ARTD1, since inhibition of ADP-ribosylation did not cause deceleration of cell cycle re-entry or G1/S progression. Thus, the cyclin E promoter state was not regulated via ADP-ribosylation of histone modifiers, such as the demethylase KDM5B,Citation38 and the levels of H3K4me3 were unchanged upon siARDT1 treatment. Regulation of transcription by ARTD1 independent of enzymatic activity has previously also been observed for IL6 in mouse and human fibroblast cells stimulated with LPS.Citation39 However, in contrast to the present study, ARTD1 suppressed IL6 expression. Thus, ARTD1 acts by different mechanisms as a co-activator or co-repressor for different types of genes, depending on the cell type and stimulus.
Beside a reduced acetylation of H4, the main observed changes in siARTD1-treated cells included an increase in H1.2 and a reduction of the H2A.Z levels at the cyclin E promoter. Increased H1 recruitment as well as decreased H2A.Z were both reported to coincide with a restrictive chromatin state and reduced gene expression.Citation36,40 While exchange of H1 by ARDT1 has previously been documented for many promoters of actively transcribed genes,Citation40 changes of H2A.Z have not been described before. It remains to be investigated whether the alterations of H2A.Z and H1 are regulated by separate mechanisms, or whether the H2A.Z changes are a consequence of the observed H1 changes. Moreover, it is not clear by which mechanism ARTD1-dependent regulation is confined to cyclin E expression, without affecting other E2F-1 target genes. It will be interesting to study whether the recruitment of ARTD1 to promoters is fine-tuned and regulated by posttranslational modifications of ARTD1 (e.g., acetylation, mono-ADP-ribosylation, phosphorylation, sumoylation or ubiquitylation), as suggested previously.Citation41
In contrast to cyclin E expression, our studies revealed that the expression levels of 2 other E2F-1-induced miRNAs (microRNAs 15 and 16) are strongly enhanced after siARTD1 treatment, suggesting that ARTD1 functions as a co-activator as well as a co-repressor within the same cell. Alternatively, the reduced levels of ARTD1 could lead to a genome-wide redistribution of transcription factors (e.g. RNA polymerase II), thereby leading to the observed altered transcriptional activation profiles. However, at the cellular level, these differential regulatory effects of reduced cyclin E expression and enhanced microRNA15/16 levels would both lead to reduced cyclin E protein levels, since the miRNAs regulate cyclin E expression negatively.Citation42 Interestingly, stably transduced shARTD1 T24 cells overcame the observed cell cycle delay already after 1-2 passages (not shown), suggesting that other cell cycle regulatory factors such as cyclin D or cyclin A could functionally compensate for the loss of cyclin E in these cells, as was speculated to be the case in cyclin E knockout mice,Citation43 or that the induced permissive chromatin changes are over-ruled by the plasticity of the chromatin (e.g., by compensatory mechanisms).
Interestingly, inhibition of ADP-ribosylation with PARP inhibitors (e.g., olaparip) did not inhibit progression of cells from G0 to G1, but rather decelerated cell cycle progression during the late S phase, indicating that ADP-ribosylation is not required for the above-described functions during T24 cell cycle re-entry or early G1/S progression, but rather affected the progression through the S phase. This observation is in contrast to earlier reports on serum-stimulated, quiescent fibroblasts and lectin-stimulated, peripheral, mononucleated blood cells.Citation25 The opposing findings thus highlight the importance of studying cellular processes in undisturbed systems, because stimulation such as by serum or lectin treatment likely induce cellular stress and consequently ADP-ribosylation. The functional contribution of ADP-ribosylation during the late S phase has already been reported in a recent publication, providing evidence that ARDT1 interacts with pRNA and TIP5 during the late S phase in an activity-dependent manner and that this interaction is important during the formation of heterochromatin.Citation28 Treatment with PARP inhibitors would thus lead to disrupted or unstable heterochromatin, which results, among other things, in sister chromatid exchange formation.
Moreover, knockdown of ARTD1 enhanced γH2AX formation, without inducing the DNA damage response as analyzed by Western blot analysis of p53 stability, replication protein A (RPA), and Chk-1 phosphorylation. It remains to be investigated how the lack of ARTD1 induces γH2AX formation in confluent T24 cells and whether this chromatin mark regulates the histone modifications observed at the cyclin E promoter.
While ARTD1 knockdown reduced cellular cyclin E protein levels, the expression of Cdk2 was even slightly increased under the same conditions. The in vitro activity of Cdk2 immunoprecipitated from siARTD1-treated cells 12 h after re-entering the cell cycle (i.e. G1 phase) was not significantly different compared to that from corresponding siMock-treated cells, suggesting that Cdk2 per se was able to form an active complex with cyclin E in siARTD1-treated cells. To exclude that Cdk2 formed a complex with other cyclins and to confirm that the immunoprecipitated Cdk2 complex indeed contains cyclin E, the cyclin E-dependent Cdk2 kinase activity should be investigated upon immunoprecipitation of cyclin E. Unforutnately, we were not able to perform cyclin E immunoprecipitation with 2 different commercially available antibodies. We can thus not completely exclude that the observed Cdk2 activity might be co-regulated by cyclin A. However, since the Cdk2 immunprecipitation was performed with lysates of cells 12 h after release into the cell cycle, which correlates with cells in G1 phase, the observed complex is more likely a cyclin E/Cdk2 complex, rather than cyclin A/Cdk2 complex.
The CIP/KIP family members p21 and p27 are highly homologous and therefore believed to function similarly,Citation44 and both proteins have been implicated in the regulation of cellular processes such as apoptosis and transcriptional activation.Citation9,45 p21 and p27 have been reported to repress transcription indirectly by inhibiting cyclin-Cdk complexes and the consecutive phosphorylation of Rb-family proteins (p107, p110, and p130). In turn, hypophosphorylated Rb-related proteins sequester E2F family members and thereby repress the transcriptional targets of this transcription factor family.Citation46 Phosphorylation of p27 at T187 is an important activity of the cyclin E/Cdk2 complex.Citation10 Cyclin E/Cdk2-dependent phosphorylation of p27 results in elimination of p27 through protein degradation, allowing cells to transit from G1 to the S phase.Citation37 Upon ARTD1 knockdown, we observed increased p27 levels 8 to 10 h after the T24 cells were allowed to re-enter the cell cycle, which could explain the observed delay in G1/S progression (). Whether the observed p27 change is mainly due to a reduced cyclin E/Cdk2 complex activity within the cells, or regulated by other mechanism (e.g. enhanced translation) has to be investigated further, however.
Together, we here describe that ARTD1 regulates the expression of cyclin E and consequently cell cycle re-entry and G1/S progression in T24 cells. The results have thus uncovered novel, non-redundant functions of ARTD1 in cell cycle regulation, which may be relevant for cancer progression.
Materials and methods
Cell culture
T24 cells were cultivated in McCoy's 5A medium (Gibco, Invitrogen, CA, California, USA) at 37°C. All media were supplemented with 1% (v/v) Penicillin/Streptavidin and 10% (v/v) fetal calf serum (Gibco, Invitrogen, CA, California, USA).
siRNA transfection
Negative control allstars (siMock), and human siPARP1 #6 were from Qiagen (Hilden, Germany). Cells were seeded 1 day before transfection (5 × 105 cells per 6 cm plate) and transfection was carried out with 40 nmol siRNA per plate and RNAi MAX lipofectamine (Invitrogen, Carlsbad, CA, USA).
Antibodies
Following antibodies were used: From Santa Cruz Biotechnology, Inc. (Dallas, TX, USA): PARP1/ARDT1 (H-250, rabbit); PCNA (PC10. mouse); p53 (FL-393,rabbit); p27 (C-19, rabbit), cyclin E (HE12, mouse), E2F-1 (C-20-rabbit), PARP-1 (C2-10, mouse), Cdk2 (D19, mouse), Rb (IF8 – mouse). From Active motif (Carlsbad, CA, USA): PARP2 (rabbit). From Sigma Aldrich (St. Louis, MO, USA): tubulin (mouse). From Millipore (MS, USA): H3K4me3 (rabbit); phospho-Histone H2A.X (mouse); H2A (rabbit); acetyl-Histone H4 (rabbit). From Abcam pls (Cambridge, UK): Histone H2A.Z (rabbit), H1.2 (rabbit); Histone H3 (rabbit). From Cell Signaling Technology, Inc. (MS, USA): P-Chk1 (S345, rabbit. From NeoMarkers (Fremont, CA, USA): RPA / p34 (9H8, mouse). From Roche AG (Basel, Switzerland): BrdU. In house production: PAR 10H (mouse). Jackson ImmunoResearch Laboratories: CyTM3-conjugated AffiniPure Goat Anti-Rabbit (Suffolk, UK). Epitomics (Burlingame, California, USA): Rb.
Cell cycle analysis by flow cytometry
Cells were harvested with trypsin and washed once with PBS. Cells (at least 3.5×10Citation5) were fixed (70% ethanol, at least 30 min on ice or overnight at 4°C), washed once with PBS and centrifuged (865 g, 4°C, 8 min). Cells were stained with propidium iodide (Sigma Aldrich, St. Louis, MO, USA), final concentration of 20 µg/ml in PBS with the addition of 100 µg/ml RNase A (37°C, 30 min in the dark). Flow cytometry analysis was performed with the CyANTM ADP 9 Analyzer (Beckman Coulter, Fullerton, CA, USA).
RNA extraction and qPCR analysis
Cells were harvested either by trypsin or directly lysed on the plate in lysis buffer. RNA extraction was performed with the NucleoSpin® RNA II kit (Macherey-Nagel, Düren, Germany). RNA was quantified with a NanoDrop (ThermoFisherScintific, Waltham, MS, USA) and reverse transcribed according to the supplier's protocol (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Foster City, CA, United States).
Quantitative-real-time polymerase chain reactions (qPCR) were performed with SYBR® green SensiMix SYBR Hi-ROX Kit (Bioline Reagents Ltd, London, UK) and a Rotor-Gene Q 2plex HRM System (Qiagen, Hilden, Germany).
Cell lysis, SDS-PAGE and western blot analysis
Whole cell lysis was performed either with trypsinized cells or directly on plates by using a Tris lysis buffer (50 mM Tris pH 8, 500 mM NaCl, 1% Triton X-100, 1 µg/ml pepstatin, 1 µg/ml bestatin, 1 µg/ml leupeptin, 2 mM PMSF; 10 min, 4°C). Bradford assay (Bio-Rad laboratories, Hercules, CA, USA) was performed and, if not indicated otherwise, 30 µg of protein extract was loaded and separated on a 10% or 12% SDS-polyacrylamide gel (120 V). The gel was blotted on a PDVF membrane and analyzed by using protein specific antibodies.
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as previously describedCitation47 by using magnetic Dynabeads® (Life Technologies, Carlsbad, CA, USA).
Cdk2-cyclin E activity assay
T24 cells were harvested at the indicated time point for immunoprecipiation. Cells were lysed in 400 µl cold lysis buffer (50 mM Tris-HCl pH 7.5, 120 mM NaCl, 0.5 mM DTT, 0.05 % NP-40, 20 mM NaF, 1 mM EDTA, 6 mM EGTA, various phosphatase inhibitors). 500 µg of protein extract was used for immunoprecipitation. The extract was incubated with the indicated antibody (1 µg) for 3 h at 4°C. Sepharose beads were added for 30 min at 4°C afterwards. The kinase activity assay was carried out in NEB3 buffer. Histone 1 (µg/µl) was added as substrate as well as radioactive ATP (32P). Phosphatase inhibitors (NPP and Na-vanadate) were added. As positive control, Cdk2-cyclinA protein was used. The reaction was carried out for 10 min at 37°C. Five µl of 6× Laemmli buffer was added and the reaction boiled at 95°C for 5 min. The extract was loaded onto a 13.5% SDS gel.
Immunofluorescence microscopy
Immunofluorescence staining for PAR in methanol/acetic acid-fixed cells was performed as described previously.Citation48
For γH2AX immunofluorescence, 45'000 T24 cells were seeded onto coverslips in 24-well plates, transfected with siRNA against siARTD1 or scrambled siRNA and grown until confluent. If required, cells were stimulated for 10 min with 1 mM H2O2 prior to being washed with PBS. Cells were fixed for 15 min with 4% formaldehyde in PBS and permeabilized for 10 min with 0.2% Triton X-100 in PBS. After blocking for 45 min in PBS containing 2% BSA and 0.1% Triton X-100, cells were stained for 1 h with the primary antibody diluted in blocking solution. Coverslips were then washed with PBS, incubated with the secondary antibody for 1 h, stained with Hoechst and mounted on glass slides using VECTASHIELD. All antibodies were used at a dilution of 1:250. Images were acquired using an inverted fluorescence microscope at 40/60x, oil immersion (Leica).
Evaluation of DNA damage
DNA damage (double-strand breaks, single-strand breaks, abasic sites) in cells was determined using the Alkaline Comet Assay (Trevigen) according to the manufacturer's instructions. %DNA in tail was calculated using Comet Assay IV (Perceptive Instruments).
Abbreviations
| ARTC | = | ADP-ribosyltransferase cholera toxin-like |
| ARTD | = | ADP-ribosyltransferase diphtheria toxin-like |
| Cdk | = | cyclin-dependent kinase |
| ChIP | = | chromatin immuno-precipitation |
| PAR | = | poly-ADP-ribose |
| PTM | = | post-translational modification |
| pRb | = | retinoblastoma protein |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
K.L. and A.H. designed and performed experiments, M.F. performed experiments, M.O.H. designed and supervised the work, and wrote the manuscript with K.L.
1195530_Supplemental_Material.zip
Download Zip (3.5 MB)Acknowledgments
We are grateful to R. Santoro (University of Zurich) for providing the BrdU antibody and for support in the course of the BrdU and ChIP experiments, to S. Ferrari (University of Zurich) for support and the Cdk2, cyclin A and cyclin B antibodies, to U. Hübscher (University of Zurich) for providing the Cdk2-cyclin A proteins, to A. Groth (University of Copenhagen, Denmark) for technical support. M. Stucki (University of Zurich) is acknowledged for providing the T24 cell line. F. Freimoser and S. Christen (University of Zurich) provided editorial assistance and critical input during the writing. This work was supported in part by Swiss National Science Foundation Grants 310030B_138667, 310030_157019, and PDMFP3_127315 (to M.O.H.), as well as the Forschungskredit from the Universität Zürich (to K.L.), and the Kanton of Zurich (to M.O.H.).
Related Research Data
References
- Murray AH, Hunt T. The cell cycle: an introduction. New York: Oxford University Press, 1993.
- Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 2000; 14:804-16; PMID:10766737
- Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004; 117:239-51; PMID:15084261; http://dx.doi.org/10.1016/S0092-8674(04)00300-9
- Rissland OS, Hong SJ, Bartel DP. MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 2011; 43:993-1004; PMID:21925387; http://dx.doi.org/10.1016/j.molcel.2011.08.021
- Sage J. Cyclin C makes an entry into the cell cycle. Dev Cell 2004; 6:607-8; PMID:15130482; http://dx.doi.org/10.1016/S1534-5807(04)00137-6
- Chatterjee SJ, George B, Goebell PJ, Alavi-Tafreshi M, Shi SR, Fung YK, Jones PA, Cordon-Cardo C, Datar RH, Cote RJ. Hyperphosphorylation of pRb: a mechanism for RB tumour suppressor pathway inactivation in bladder cancer. J Pathol 2004; 203:762-70; PMID:15221935; http://dx.doi.org/10.1002/path.1567
- Cobrinik D. Pocket proteins and cell cycle control. Oncogene 2005; 24:2796-809; PMID:15838516; http://dx.doi.org/10.1038/sj.onc.1208619
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev 1998; 12:2245-62; PMID:9694791; http://dx.doi.org/10.1101/gad.12.15.2245
- Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 2003; 13:65-70; PMID:12559756; http://dx.doi.org/10.1016/S0962-8924(02)00043-0
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997; 11:1464-78; PMID:9192873; http://dx.doi.org/10.1101/gad.11.11.1464
- Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 1999; 274:17860-8; PMID:10364231; http://dx.doi.org/10.1074/jbc.274.25.17860
- Kanai M, Uchida M, Hanai S, Uematsu N, Uchida K, Miwa M. Poly(ADP-ribose) polymerase localizes to the centrosomes and chromosomes. Biochem Biophys Res Commun 2000; 278:385-9; PMID:11097846; http://dx.doi.org/10.1006/bbrc.2000.3801
- Saxena A, Wong LH, Kalitsis P, Earle E, Shaffer LG, Choo KH. Poly(ADP-ribose) polymerase 2 localizes to mammalian active centromeres and interacts with PARP-1, Cenpa, Cenpb and Bub3, but not Cenpc. Hum Mol Genet 2002; 11:2319-29; PMID:12217960; http://dx.doi.org/10.1093/hmg/11.19.2319
- Saxena A, Saffery R, Wong L, Kalitsis P, Choo K. Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated. J Biol Chem 2002; 277:26921-6; PMID:12011073; http://dx.doi.org/10.1074/jbc.M200620200
- Monaco L, Kolthur-Seetharam U, Loury R, Murcia JM, de Murcia G, Sassone-Corsi P. Inhibition of Aurora-B kinase activity by poly(ADP-ribosyl)ation in response to DNA damage. Proc Natl Acad Sci USA 2005; 102:14244-8; PMID:16179389; http://dx.doi.org/10.1073/pnas.0506252102
- Caiafa P, Guastafierro T, Zampieri M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J 2009; 23:672-8; PMID:19001527; http://dx.doi.org/10.1096/fj.08-123265
- Wesierska-Gadek J, Schloffer D, Gueorguieva M, Uhl M, Skladanowski A. Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout cells to antitumor triazoloacridone C-1305 is associated with permanent G2 cell cycle arrest. Cancer Res 2004; 64:4487-97; PMID:15231658; http://dx.doi.org/10.1158/0008-5472.CAN-03-3410
- Tanuma S, Kanai Y. Poly(ADP-ribosyl)ation of chromosomal proteins in the HeLa S3 cell cycle. J Biol Chem 1982; 257:6565-70; PMID:7042716
- Tentori L, Muzi A, Dorio AS, Scarsella M, Leonetti C, Shah GM, Xu W, Camaioni E, Gold B, Pellicciari R, et al. Pharmacological inhibition of poly(ADP-ribose) polymerase (PARP) activity in PARP-1 silenced tumour cells increases chemosensitivity to temozolomide and to a N3-adenine selective methylating agent. Curr Cancer Drug Targets 2010; 10:368-83; PMID:20464779; http://dx.doi.org/10.2174/156800910791208571
- Kashima L, Idogawa M, Mita H, Shitashige M, Yamada T, Ogi K, Suzuki H, Toyota M, Ariga H, Sasaki Y, et al. CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J Biol Chem 2012; 287:12975-84; PMID:22337872; http://dx.doi.org/10.1074/jbc.M111.321828
- Wright RH, Castellano G, Bonet J, Le Dily F, Font-Mateu J, Ballare C, Nacht AS, Soronellas D, Oliva B, Beato M. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev 2012; 26:1972-83; PMID:22948662; http://dx.doi.org/10.1101/gad.193193.112
- Madison DL, Lundblad JR. C-terminal binding protein and poly(ADP)ribose polymerase 1 contribute to repression of the p21(waf1/cip1) promoter. Oncogene 2010; 29:6027-39; PMID:20711239; http://dx.doi.org/10.1038/onc.2010.338
- Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, Hottiger MO, Smulson ME. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 2003; 22:8460-71; PMID:14627987; http://dx.doi.org/10.1038/sj.onc.1206897
- Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Smulson ME. Poly(ADP-ribose) polymerase upregulates E2F-1 promoter activity and DNA pol alpha expression during early S phase. Oncogene 1999; 18:5015-23; PMID:10490838; http://dx.doi.org/10.1038/sj.onc.1202900
- Carbone M, Rossi MN, Cavaldesi M, Notari A, Amati P, Maione R. Poly(ADP-ribosyl)ation is implicated in the G0-G1 transition of resting cells. Oncogene 2008; 27:6083-92; PMID:18663363; http://dx.doi.org/10.1038/onc.2008.221
- Chang P, Jacobson MK, Mitchison TJ. Poly(ADP-ribose) is required for spindle assembly and structure. Nature 2004; 432:645-9; PMID:15577915; http://dx.doi.org/10.1038/nature03061
- Chang P, Coughlin M, Mitchison TJ. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat Cell Biol 2005; 7:1133-9; PMID:16244666; http://dx.doi.org/10.1038/ncb1322
- Guetg C, Scheifele F, Rosenthal F, Hottiger MO, Santoro R. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol Cell 2012; 45:790-800; PMID:22405650; http://dx.doi.org/10.1016/j.molcel.2012.01.024
- Horton J, Stefanick D, Naron J, Kedar P, Wilson S. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem 2005; 280:15773-85; PMID:15701627; http://dx.doi.org/10.1074/jbc.M413841200
- Bäckert S. Involvement of PARP1 in NF-κB-dependent gene expression during the cell cycle. University of Zurich, Vetsuisse Faculty, 2009. http://dx.doi.org/10.5167/uzh-32472 (D.V.M. thesis)
- Peng CC, Chen KC, Peng RY, Su CH, Hsieh-Li HM. Human urinary bladder cancer T24 cells are susceptible to the Antrodia camphorata extracts. Cancer Lett 2006; 243:109-19; PMID:16455193; http://dx.doi.org/10.1016/j.canlet.2005.11.021
- Cooper MJ, Haluschak JJ, Johnson D, Schwartz S, Morrison LJ, Lippa M, Hatzivassiliou G, Tan J. p53 mutations in bladder carcinoma cell lines. Oncol Res 1994; 6:569-79; PMID:7787250
- Jin Y, Xu X, Yang M, Wei F, Ayi T, Bowcock A, Baer R. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc Natl Acad Sci USA 1997; 94:12075-80; PMID:9342365; http://dx.doi.org/10.1073/pnas.94.22.12075
- Chen Y, Farmer AA, Chen CF, Jones DC, Chen PL, Lee WH. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res 1996; 56:3168-72; PMID:8764100
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A 1995; 92:12146-50; PMID:8618861; http://dx.doi.org/10.1073/pnas.92.26.12146
- Ku M, Jaffe JD, Koche RP, Rheinbay E, Endoh M, Koseki H, Carr SA, Bernstein BE. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol 2012; 13:R85; PMID:23034477; http://dx.doi.org/10.1186/gb-2012-13-10-r85
- Garcia-Bassets I, Kwon Y-S, Telese F, Prefontaine G, Hutt K, Cheng C, Ju B-G, Ohgi K, Wang J, Escoubet-Lozach L, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007; 128:505-18; PMID:17289570; http://dx.doi.org/10.1016/j.cell.2006.12.038
- Krishnakumar R, Kraus W. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 2010; 39:736-49; PMID:20832725; http://dx.doi.org/10.1016/j.molcel.2010.08.014
- Minotti R, Andersson A, Hottiger MO. ARTD1 Suppresses Interleukin 6 Expression by Repressing MLL1-Dependent Histone H3 Trimethylation. Mol Cell Biol 2015; 35:3189-99; PMID:26149390; http://dx.doi.org/10.1128/MCB.00196-15
- Krishnakumar R, Gamble M, Frizzell K, Berrocal J, Kininis M, Kraus W. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008; 319:819-21; PMID:18258916; http://dx.doi.org/10.1126/science.1149250
- Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26:417-32; PMID:22391446; http://dx.doi.org/10.1101/gad.183509.111
- Ofir M, Hacohen D, Ginsberg D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res 2011; 9:440-7; PMID:21454377; http://dx.doi.org/10.1158/1541-7786.MCR-10-0344
- Lents NH, Baldassare JJ. CDK2 and cyclin E knockout mice: lessons from breast cancer. Trends Endocrinol Metab 2004; 15:1-3; PMID:14693416; http://dx.doi.org/10.1016/j.tem.2003.10.011
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996; 382:325-31; PMID:8684460; http://dx.doi.org/10.1038/382325a0
- Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 2001; 264:148-68; PMID:11237531; http://dx.doi.org/10.1006/excr.2000.5143
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/10.1101/gad.13.12.1501
- Santoro R, Li J, Grummt I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 2002; 32:393-6; PMID:12368916; http://dx.doi.org/10.1038/ng1010
- Bartolomei G, Leutert M, Manzo M, Baubec T, Hottiger MO. Analysis of Chromatin ADP-Ribosylation at the Genome-wide Level and at Specific Loci by ADPr-ChAP. Mol Cell 2016; 61:474-85; PMID:26833088; http://dx.doi.org/10.1016/j.molcel.2015.12.025