
ABSTRACT
The epithelial-mesenchymal transition (EMT) is thought to be essential for cancer metastasis. While chromatin remodeling is involved in EMT, which processes contribute to this remodeling remain poorly investigated. Recently, we showed that silencing or removal of the histone variant H2A.X induced mesenchymal-like characteristics, including activation of the EMT transcription factors, Slug and Zeb1 in human colon cancer cells. Here, we provide the evidence that H2A.X loss in human non-tumorigenic breast cell line MCF10A results in a robust EMT activation, as substantiated by a genome-wide expression analysis. Cells deficient for H2A.X exhibit enhanced migration and invasion, along with an activation of a set of mesenchymal genes and a concomitant repression of epithelial genes. In the breast model, the EMT-related transcription factor Twist1 cooperates with Slug to regulate EMT upon H2A.X Loss. Of interest, H2A.X expression level tightly correlates with Twist1, and to a lesser extent with Slug in the panel of human breast cancer cell lines of the NCI-60 datasets. These new findings indicate that H2A.X is involved in the EMT processes in cells of different origins but pairing with transcription factors for EMT may be tissue specific.
KEYWORDS:
Introduction
The vast majority of deaths from cancer are due to metastatic disease.Citation1 How to treat cancers arising from the spread of cancer cells to distant organs is a longstanding clinical challenge. Cells from primary tumors gain an invasive phenotype through a process widely known as epithelial-mesenchymal transition (EMT). EMT is characterized by the loss of cell-to-cell adhesion, disruption of the apicobasal polarity and transition to a cell type with increased spindle-like morphology. The molecular mechanisms underlying EMT include decreased expression of epithelial genes with the concomitant activation of mesenchymal genes, the expression of matrix metalloproteinases markers, and the formation of lamellipodia, filopodia and invadopodia.Citation2 In turn, these changes allow cells to invade the extracellular matrix.Citation3 In a reverse transition, MET, mesenchymal cells may revert to the epithelial phenotype and may be involved in the establishment of tumors at distant sites. EMT involves several master regulators, namely the EMT transcription factors which consist of a family of genes including Snail (Snail1 and Snail2), Twist (Twist1 and Twist2) and zinc-finger E-box-binding Zeb (Zeb1 and Zeb2) among others. These transcription factors are expressed early in EMT, and are reported to play central roles in the development of cancers as well as in fibrotic diseases.Citation2,4,5 Since Snails, Twists and Zebs display distinct expression profiles, it has been accepted that their contributions to EMT depend on the cell or tissue type and the signaling pathways upstream to EMT activation. Moreover, EMT transcription factors control the expression of each other, and to some extent they cooperate to drive EMT program activation.Citation6,7
Changes in the chromatin configuration have emerged as key players in EMT-related transcription factor regulation,Citation8 including chromatin modifications which enable the reprogramming of gene expression during EMT. For instance, the cellular mechanism for epithelial gene repression depends on relative modifications of the bivalent marks H3K4me3 (active transcription)/ H3K27me3 (gene silencing), with a gain of histone H3K27me3, and loss of H3K4me3 toward repression.Citation9 DNA methyltransferases catalyzing gene silencing of promoters of genes that confer epithelial traits, are also involved in EMT.Citation10,11 Thus, changes in the epigenetic modifications between H3K4/K27me3 or H3K4me3/DNAme result in an alteration of gene expression and EMT.
While epigenetic changes are accepted as an integral part of the EMT, whether changes in the structure of the core nucleosome itself affect the regulation of EMT-related genes is still poorly investigated. Recently, we showed that removal of the histone variant H2A.X, known for its role in DNA double-strand break repair and genome stability, leads to enhanced expression of the EMT-related transcription factors Slug and Zeb1, thus favoring the activation of EMT in human colon cancer cells. Whether this regulation is a broader phenomenon in epithelial cells remains unclear. Here, we provide new evidence that H2A.X loss in the human non-tumorigenic breast cell line MCF10A results in a robust EMT program activation. This program seems to involve mainly Twist1 and to a lesser extent Slug, in contrast to Zeb1 and Slug in colon cancer cells. These results are substantiated by the evidence that the co-silencing of Twist1 and Slug leads to a significant reversion of EMT marker expression. Our new evidence suggests that H2A.X-driven EMT occurs in epithelial cells of different origins, howerer, different sets of EMT-related transcription factors appear to be involved.
Results and discussion
H2A.X removal leads to epithelial-mesenchymal transition in MCF10A cells
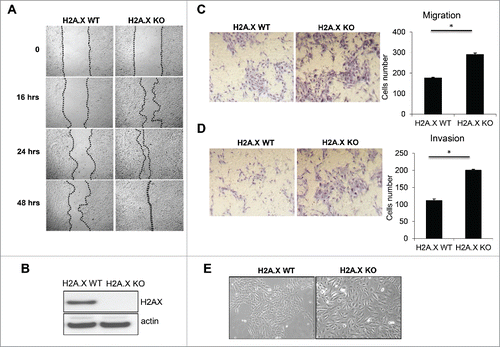
In a recent study, we reported that when the human colon cancer line HCT116 was made deficient for the histone variant H2A.X, the cells acquired invasive properties and exhibited enhanced expression of several mesenchymal genes coupled with reduced expression of many epithelial markers.Citation12 In the present study, we asked whether epithelial cells from a different origin, breast tissue, might also exhibit EMT following H2A.X removal. Utilizing normal and H2A.X-null MCF10A cell lines, we observed that the H2A.X deletion correlated with elevated levels of migration and invasion, as substantiated by wound healing and transwell invasion assays respectively (). H2A.X-null MCF10A cells exhibited enhanced wound healing ability (), and also harbored stronger migration and invasion potential (). Of interest, H2A.X knockout cells exhibited enlarged and scattered morphology (), consistent with the phenotype of cells undergoing EMT.Citation13,14 Taken together, these data indicate that H2A.X removal in MCF10A human breast cells results in the activation of EMT program similarly to H2A.X removal in the human colon cancer line HCT116.
Figure 1. H2A.X loss leads to increased migration and invasion. (A) MCF10A parental and H2A.X knockout cells were cultured for 48 hours (90–95% confluence) prior to a scratch assay. A 200 µl pipet tip was then used to create a wound and cells were followed up for indicated times. Representative phase contrast images were recorded at 10x magnification. (B) Immunoblot of MCF10A parental and H2A.X knockout cells. (C-D) H2A.X knockout cells exhibit increased migration (C) and invasion (D) in transwell invasion assays. Quantification are shown in the right panels. (E) Photomicrographs of MCF10A parental and H2A.X knockout cells.
Differential gene expression analysis
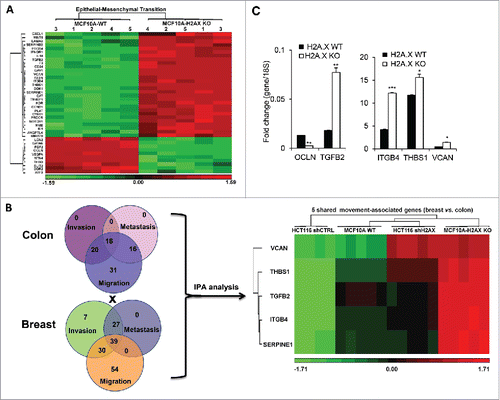
We then performed a genome-wide differential gene expression analysis comparing the human breast cell line MCF10A H2A.X-knockout and parental cells. (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=kfszmawszlizlcbandacc=GSE80180). The output from Ingenuity Pathway Analysis (IPA) using the 2315 Differentially Expressed Genes (DEGs) revealed 39 genes involved in Epithelial-Mesenchymal Transition, with some of these genes associated with movement-related phenotypes, namely cell invasion, migration and metastasis (). Many of these genes including VCAN, THBS1 and ITGB4 have been widely studied for their key roles in metastasis and EMT. For instance, a report using a model of spontaneous breast cancer showed that bone marrow-derived myeloid progenitor cells secreting the proteoglycan versican (VCAN) correlated with accelerated formation of lung metastases.Citation15 Thrombospondin 1 (THBS1) and integrin β4 (ITGB4) have also been described as promoters of thyroid cancer progression and the invasive potential of gastric cancer cells respectively.Citation16,17 Additional genes including WNT5, NOTCH1, CAV1 and CCND1 have also been observed in several models of EMT and metastasis.Citation4,18-21 These observations indicate that H2A.X loss leads to the activation of EMT program in breast cells. As many of the movement-associated genes observed in MCF10A cells were also found to regulate EMT, migration and invasion in human colon cancer cell line HCT116,Citation12 our data suggest the existence of H2A.X-driven EMT signature shared by breast and colon cancer cells. We then analyzed the movement-associated genes shared between MCF10A and HCT116 cells. Data revealed VCAN, THBS1, TGFB2, ITGB4 and SERPINE1 as genes commonly overexpressed in cells deficient for H2A.X (). Further validation was made using real time PCR quantification to access the transcript levels of these genes with the inclusion of occludin (OCLN) as one of the key epithelial gene found to be downregulated in MCF10A H2A.X knockout cells. Data indicate that H2A.X loss promotes increased expression of VCAN, THBS1, TGFB2, ITGB4, while repressing OCLN expression (). These data raised the important question whether these genes are conserved in the pathways associated with H2A.X loss-induced EMT in epithelial cells. The loss of epithelial phenotypes and the acquisition of mesenchymal traits through EMT activation have been shown to enhance the invasive properties of epithelial cells in a variety of cancer models including breast cancer.Citation22
Figure 2. H2A.X loss in MCF10A results in a robust activation of Epithelial-Mesenchymal Transition. (A) Heat map of the 39 differentially expressed genes (DEGs) related to Epithelial-Mesenchymal Transition (EMT). Five (5) replicates were used for either parental or H2A.X Knockout cells. (B) (Left) Venn diagrams of differentially expressed genes in human colon cancer cells HCT116 and breast cells MCF10A, associated with the invasion, migration and metastasis pathways using IPA. Differentially expressed genes (DEGs) were generated using Partek Genomics Suite software, version 6.6. (Right) Heat map of the 5 movement-associated genes shared by colon cancer cells HCT116 and breast cells MCF10A. (C) Verification of the main genes shared in (B) by real time PCR. Expression values are relative fold change for gene transcripts normalized to 18S RNA (gene/18S ratio). Error bars represent the SEM (n = 3). Statistical significance was determined by a 2-tailed, unpaired Student's t-test.
Twist1 cooperates with slug to control H2A.X loss-induced EMT in MCF10A cells
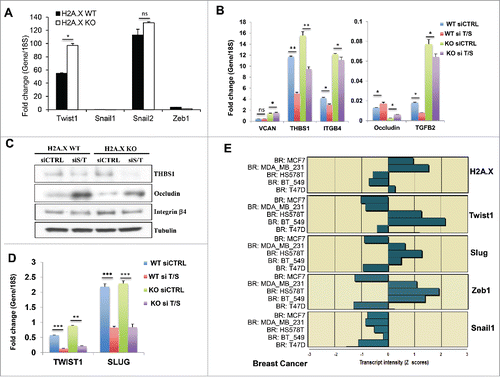
Since H2A.X-driven EMT exhibits similar changes in breast and colon cancer cells, we compared the transcriptional factors regulating VCAN, THBS1, TGFB2, ITGB4 and SERPINE1 in the 2 lines. EMT is activated through a set of pleiotropically acting transcription factors, including Snail1/2,Citation23 Zeb1/2,Citation24,25 Twist1/2Citation26 FOXC2Citation27 among others.Citation2 It is widely accepted that the E-box-binding zinc finger transcription factors Slug and Zeb1 contribute to the EMT in primary epithelial cancers cells.Citation4 However Twist1/2 and other EMT-related transcription factors may be essential in other EMT models. In our recent study, we reported that Slug (Snail2) and Zeb1 concomittantly regulate H2A.X loss-induced EMT in human colon cancer cells.Citation12 As none of these EMT transcription factors was found in our micro array data sets from MCF10A cells, we accessed the transcript levels of the main EMT transcription factors including Twist1, Snail1, Snail2 (Slug) and Zeb1 using real time PCR assay. Our data indicated that in contrast to HCT116 cells,Citation12 Twist1 was found overexpressed in H2A.X-deficient MCF10A cells (), but with only a modest elevation in Slug expression. Of interest, Twist1 was described to be a master regulator of EMT in several breast epithelial cells including MCF10A.Citation28-30 To determine whether these transcription factors are essential in H2A.X loss-induced EMT in breast cells, we performed a transcient silencing of Twist1 and Slug using small interference RNA. While an independent knockdown of Twist failed to significantly affect most of the EMT markers analyzed (data not shown), the concomitant silencing of Twist1 and Slug resulted in varied levels of reversion of THBS1, TGFB2, ITGB4 and OCLN transcript and protein (), whereas the transcript level of VCAN remains unchanged. This partial EMT reversal most importantly raises the important question whether H2A.X loss induced-EMT requires additional EMT transcription factors in breast cells. Nevertheless, these data indicate that H2A.X-driven EMT occurs in epithelial cells of different origins, although the mechanism may involve different set of EMT-related transcription factors. Furthermore, utilizing the NCI60 genome-wide expression dataset (http://discover.nci.nih.gov/cellminer/), we found a strong negative correlation between H2A.X and Twist1 expression in all of the 5 breast cancer cell lines of the data sets (). The correlation was lower for Slug and Zeb1 while no correlation was found for Snail1(). These observations suggested that H2AX loss-induced EMT in breast cancer cells utilized an hierarchical cluster of EMT transcription factors in which Twist1 may play an essential role.
Figure 3. The EMT-related transcription factors Twist1 and Slug mediate in H2A.X loss-induced EMT. (A) Analysis of the transcript levels of Twist1, Snail1, Snail2 (Slug) and Zeb1 in MCF10A cells. Expression values are relative fold change for gene transcripts normalized to 18S RNA (gene/18S ratio). Error bars represent the SEM (n = 3). Statistical significance was determined by a 2-tailed, unpaired Student's t-test. (B) Effects of Twist1 and Slug co-silencing on the transcript levels of VCAN, THBS1, ITGB4, OCLN (occluding) and TGFB2. Cells were transfected for 3 days with control siRNA (siC) or with a pool of siTwist1 and siSlug (siT/S). Expression values are relative fold change for gene transcripts normalized to 18S RNA (gene/18S ratio). It should be noticed that this panel is an extension of the , with inclusion of the siTwist/Slug transcript values. The two panels are presented separately to allow smooth reading of this report. Error bars represent the SEM (n = 3). Statistical significance was determined by a 2-tailed, unpaired Student's t-test. (C) Immunoblot of THBS1, ITGB4 (integrin β4) and OCLN (occluding) upon Twist1 and Slug co-silencing. Cells were transfected for 3 days with control siRNA (siC) or with a pool of siSlug and siTwist1 (siS/T). Immunoblot analysis was performed using tubulin as loading control. (D) Verification of the Twist1 and Slug transcript levels by real time PCR. Cells were transfected for 3 days with control siRNA (siC) or with a pool of siTwist1 and siSlug (siT/S). Expression values are relative fold change for gene transcripts normalized to 18S RNA (gene/18S ratio). Error bars represent the SEM (n = 3). Statistical significance was determined by a 2-tailed, unpaired Student's t-test. (E) Inverse correlation between the transcript levels of H2A.X and the EMT-related transcription factors Twist1, Slug, Zeb1 and Snail1 in the breast cancer subpanel of the NCI60. The Z score represents the transcript expression in standard deviation units relative to the gene-specific mean expression level.
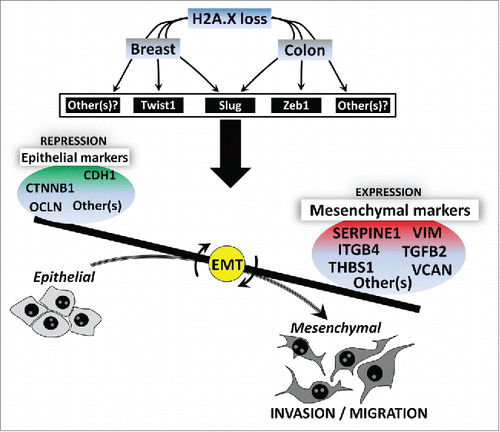
Overally, our present study indicates that H2AX loss leads to the activation of EMT program in breast cells through a set of key mesenchymal genes including VCAN, THBS1, TGFB2, ITGB4, SERPINE1 (). These genes seem to be conserved in colon cancer cells,Citation12 thus suggesting that the role of H2A.X in EMT is more broad. EMT occurs in embryogenesis and organ development, in tissue regeneration and in metastasis during cancer progression. The evidence that H2AX removal induces EMT in a non-tumorigenic (but immortalized) breast cell line further indicates that the role of H2A.X in EMT may also be a widely occurring phenomenon, not limited to cancer progression. However, it should be noticed that while Zeb1 and Slug mediate H2A.X-driven EMT in colon, Twist1 and Slug seems to be essential in breast (). Additional EMT-related transcription factors may encounter for the role of H2A.X in EMT. Since H2A.X removal leads to a selective activation of some EMT genes in relation to the microenvironmental context, investigating the role of EMT inducers such as inflammatory cytokines, oncogenic/metabolic stressors, hypoxia and miRNAsCitation31-34 in H2A.X gene regulation will provide a broader view on the role of H2A.X in EMT.
Figure 4. Hypothetical model for the role of H2A.X in EMT. H2AX loss leads to the activation of EMT program in epithelial cells (colon and breast cells) through a set of key EMT-related transcription factors. Slug and Zeb1 are prefenrentially activated in colon cells whereas Twist1 and Slug seem to be critical in breast cells. Additional EMT-related transcription factors may also encounter for the role of H2A.X in EMT. Upon H2A.X loss, cells undergo phenotypic changes through the upregulation of key mesenchymal genes including VCAN, THBS1, TGFB2, ITGB4, SERPINE1, while several epithelial genes including CDH1, CTNNB1, OCLN among others are repressed. Cells then acquire increased ability for migration and invasion.
Methods
Cell culture
Human MCF10A parental and H2A.X knockout cells were obtained from Sigma, St. Louis, MO. Cells were maintained in a special medium (Lonza, Walkersville, MD USA). The medium is provided with a cocktail of hydrocortisone, EGF, cholera toxin, insulin, and penicillin-streptomycin (Invitrogen). All cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37 C.
SiRNA transfection
Small RNA interference (siRNA) transfection experiments were performed with Dharmafect (Thermo Scientific, Hudson, NH, USA). A SMARTpool consisting of 4 short sequences of siRNA specific for Slug and Twist1 (Dharmacon, Thermo Scientific, Hudson, NH, USA) along with Scrambled control (nontargeting siRNA) were used under conditions specified by the manufacturer.
Transwell migration and invasion assays
MCF10A cells were cultured in EGF-free medium for 16 hours prior to migration and invasion assay. Matrigel inserts were hydrated for 2 hours in 500 μL serum-free DMEM; then suspensions of 2.5 × 104 cells were seeded in triplicate into each well of the 24-well plate format BD BioCoat Control and Matrigel Invasion Chambers (BD Biosciences) and incubated for 48 hours at 37°C in 5% CO2. Enriched media containing EGF was used as chemoattractant. Membranes were subsequently fixed with methanol, stained with crystal violet, and cells present on the underside of the membrane were counted.
Wound closure assay
MCF10A cells were seeded on 6-well tissue culture plates (5×105 cells/ well) 24 to 48 hrs before the wounding procedure. Wounding was performed by scratching cell monolayers with a 200 μl plastic pipette tip. Images of scratched monolayers were monitored under a phase contrast microscope.
Real-time PCR
Total RNA was extracted from cells using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Quality of RNA preparation, based on the 28S/18S rRNAs ratio, was assessed using the RNA 6000 Nano Lab-On-chip (Agilent Technologies, Palo Alto, CA, USA). Reverse transcription and Real-time PCR were performed as previously described.Citation12 Oligonucleotides were purchased from Life Technologies and their sequences are available on demand.
Microarray
Total RNAs from parental cells (MCF10A-WT) or H2AX knockout cells (MCF10A-H2AX KO) were extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) following manufacturer's instructions. All RNAs were QC-tested and used at LMT/Affymetrix Group, NCI-Frederick, MD, to perform Affymetrix GeneChip Human Gene 2.0 ST array (n = 5 for each group). Affymetrix Expression Console (EC) was used to generate CHP files from CEL files. Then, the CEL files were loaded into Partek Genomics Suite software, version 6.6 for normalization and differential expression analysis. Biological interpretation was performed with the Ingenuity Pathway Analysis (IPA) tool.
Western blots
Cells were washed twice with PBS, directly solubilized in denaturing sample buffer and then subjected to SDS-PAGE. Proteins were electrotransferred to 0.2 µm Protan BA 83 nitrocellulose sheets (Invitrogen, Carlsbad, CA, USA) for immunodetection with the following primary antibodies: H2AX (1:2000; ab20669, Abcam, Cambridge, MA, USA); ); Integrin β4 (1:1000; sc-9090, Santa Cruz Biotechnology, Santa Cruz, CA, USA); Occludin (1:500, sc-8144, Santa Cruz); THBS1 (1:1000; ab1823, Abcam, Cambridge, MA, USA); Tubulin (1:2000; ab6046, Abcam, Cambridge, MA, USA); and actin (1:2000; A5060, Sigma Aldrich, Saint Louis, MO, USA). Immune complexes were detected with horseradish peroxidase coupled anti-rabbit IgG antibody (Amersham™, GE Healthcare, Pittsburgh, PA, USA).
Statistical analysis
Parametric data were analyzed using a 2-tailed t-test. A value of P < 0.05 was considered statistically significant. Data are presented as mean ± SD.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
U.W., C.E.R. and W.M.B. designed experiments. U.W., A.S.B. and T.K.S performed experiments and analyzed data. U.W., M. K., N.A., A.M. and P.J. performed microarray analyses. U.W., C.E.R. and W.M.B wrote the manuscript.
Accession codes
Microarray data have been deposited in th the NCBI/GEO database under the accession code GSE80180.
Acknowledgments
We are indebted to members of LMT/Affymetrix Group, NCI-Frederick, MD, for their help with microarray runs (Affymetrix GeneChip Human Gene 2.0 ST array). We thank Dr. Myriem Boufraqech of Endocrine Oncology Branch (EOB/NCI) for her help with experiments.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases, Radiation/Nuclear Countermeasures Program and the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health.
References
- Coghlin C, Murray GI. Current and emerging concepts in tumour metastasis. J Pathol 2010; 222(1):1-15; PMID:20681009; http://dx.doi.org/10.1002/path.2727
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15(3):178-96; PMID:24556840; http://dx.doi.org/10.1038/nrm3758
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139(5):871-90; PMID:19945376; http://dx.doi.org/10.1016/j.cell.2009.11.007
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119(6):1420-8; PMID:19487818; http://dx.doi.org/10.1172/JCI39104
- Nistico P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol 2012; 4(2):a011908; PMID:22300978; http://dx.doi.org/10.1101/cshperspect.a011908
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7(6):415-28; http://dx.doi.org/10.1038/nrc2131
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013; 13(2):97-110; PMID:23344542; http://dx.doi.org/10.1038/nrc3447
- Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 2013; 19(11):1438-49; PMID:24202396; http://dx.doi.org/10.1038/nm.3336
- Ke XS, Qu Y, Cheng Y, Li WC, Rotter V, Øyan AM, Kalland KH. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics 2010; 11:669; PMID:21108828; http://dx.doi.org/10.1186/1471-2164-11-669
- Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, Matei D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014; 9(11):1461-72; PMID:25470663; http://dx.doi.org/10.4161/15592294.2014.971608
- Huangyang P, Shang Y. Epigenetic regulation of epithelial to mesenchymal transition. Curr Cancer Drug Targets 2013; 13(9):973-85; PMID:24168185; http://dx.doi.org/10.2174/15680096113136660103
- Weyemi U, Redon CE, Choudhuri R, Aziz T, Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, et al. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat Commun 2016; 7:10711; PMID:26876487; http://dx.doi.org/10.1038/ncomms10711
- Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005; 24(50):7443-54; PMID:16288291; http://dx.doi.org/10.1038/sj.onc.1209091
- Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4(8):657-65; PMID:12923528; http://dx.doi.org/10.1038/nrm1175
- Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res 2012; 72(19):4883-9; PMID:23002209; http://dx.doi.org/10.1158/0008-5472.CAN-12-1223
- Nucera C, Porrello A, Antonello ZA, Mekel M, Nehs MA, Giordano TJ, Gerald D, Benjamin LE, Priolo C, Puxeddu E, et al. B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc Natl Acad Sci U S A 2010; 107(23):10649-54; PMID:20498063; http://dx.doi.org/10.1073/pnas.1004934107
- Yang ZY, Jiang H, Qu Y, Wei M, Yan M, Zhu ZG, Liu BY, Chen GQ, Wu YL, Gu QL. Metallopanstimulin-1 regulates invasion and migration of gastric cancer cells partially through integrin beta4. Carcinogenesis 2013; 34(12):2851-60; PMID:23803695; http://dx.doi.org/10.1093/carcin/bgt226
- Bo H, Zhang S, Gao L, Chen Y, Zhang J, Chang X, Zhu M. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer 2013; 13:496; PMID:24156409; http://dx.doi.org/10.1186/1471-2407-13-496
- Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, Aranda S, Palomo S, McCormick F, Izpisúa-Belmonte JC, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18(1):99-115; PMID:14701881; http://dx.doi.org/10.1101/gad.276304
- Bailey KM, Liu J. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J Biol Chem 2008; 283(20):13714-24; PMID:18332144; http://dx.doi.org/10.1074/jbc.M709329200
- Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol 2014; 16(6):488-94; PMID:24875735; http://dx.doi.org/10.1038/ncb2976
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14(6):818-29; PMID:18539112; http://dx.doi.org/10.1016/j.devcel.2008.05.009
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2(2):84-9; PMID:10655587; http://dx.doi.org/10.1038/35000034
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005; 24(14):2375-85; PMID:15674322; http://dx.doi.org/10.1038/sj.onc.1208429
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 2001; 7(6):1267-78; PMID:11430829; http://dx.doi.org/10.1016/S1097-2765(01)00260-X
- El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, Csizmadia E, Mariani O, Zhu C, Campagne A, et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A 2012; 109(43):17460-5; PMID:23033492; http://dx.doi.org/10.1073/pnas.1206653109
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133(4):704-15; PMID:18485877; http://dx.doi.org/10.1016/j.cell.2008.03.027
- Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol 2015; 17(5):678-88; PMID:25893917; http://dx.doi.org/10.1038/ncb3157
- Nairismagi ML, Vislovukh A, Meng Q, Kratassiouk G, Beldiman C, Petretich M, Groisman R, Füchtbauer EM, Harel-Bellan A, Groisman I. Translational control of TWIST1 expression in MCF-10A cell lines recapitulating breast cancer progression. Oncogene 2012; 31(47):4960-6; PMID:22266852; http://dx.doi.org/10.1038/onc.2011.650
- Ai L, Kim WJ, Alpay M, Tang M, Pardo CE, Hatakeyama S, May WS, Kladde MP, Heldermon CD, Siegel EM, et al. TRIM29 suppresses TWIST1 and invasive breast cancer behavior. Cancer Res 2014; 74(17):4875-87; PMID:24950909; http://dx.doi.org/10.1158/0008-5472.CAN-13-3579
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5):646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P, Kim D, Kanarek N, Pacold ME, Chen WW, et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014; 158(5):1094-109; PMID:25171410; http://dx.doi.org/10.1016/j.cell.2014.07.032
- Wakefield LM, Hill CS. Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 2013; 13(5):328-41; PMID:23612460; http://dx.doi.org/10.1038/nrc3500
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011; 145(6):926-40; PMID:21663795; http://dx.doi.org/10.1016/j.cell.2011.04.029