
ABSTRACT
Upregulation of RNA Polymerase (Pol I)-mediated transcription of rRNA and increased ribogenesis are hallmarks of breast cancer. According to several datasets, including The Cancer Genome Atlas (TCGA), amplification/upregulation of genes encoding for basal components of the Pol I transcriptional machinery is frequent at different breast cancer stages. Here we show that knock down of the RNA polymerase I-specific transcription initiation factor RRN3 (TIF-IA) in breast cancer cells is sufficient to reduce rRNA synthesis and inhibit cell proliferation, and second that stable ectopic expression of RRN3 in human mammary epithelial (HME1) cells, by increasing rRNA transcription, confers increased sensitivity to the anti-proliferative effects of a selective Pol I inhibitor. Further, RRN3-overexpressing HME1 cells, when grown in in vitro 3-dimensional (3D) culture, develop into morphologically aberrant acinar structures lacking a lumen and filled with proliferative cells, thus acquiring a morphology resembling in situ ductal breast cancer lesions (DCIS). Consequently, interference with RRN3 control of Pol I transcription seems capable of both compromising mammary epithelial morphogenetic processes at early breast cancer stages, and driving breast cancer progression by fostering proliferation.
Introduction
RNA Polymerase I (Pol I)-mediated transcription of ribosomal genes (rDNA) into rRNA is a major rate-limiting step of ribosome biogenesis. Pol I-mediated rRNA synthesis and ribogenesis, being tightly coupled with cell growth and proliferation, have a major impact on cell fate. rRNA synthesis is downregulated when normal cells transition from a proliferative state to a growth-arrested state during terminal differentiation, while it is aberrantly upregulated in cells that evade the growth-inhibitory/differentiation action of physiological signals, as it happens in cancer.Citation1,2 Even if it is well established that increased rRNA synthesis, which is required for ribogenesis, is pivotal for cancer cell proliferation, it is not clear whether it is a cause or just a consequence of malignant cell transformation.Citation3-5
Increased rRNA synthesis and ribogenesis in breast cancer were initially inferred from the enlargement of silver-stained nucleolar regions called argyrophilic nucleolar organizer regions (AgNORs).Citation6 The overall AgNOR area significantly increases with tumor grade and correlates with a poor prognosis.Citation6,7 Remarkably, enlarged AgNORs have also been detected in benign breast lesions and in situ breast carcinoma, such as ductal carcinoma in situ (DCIS),Citation6,7 suggesting that increased rRNA synthesis and ribogenesis can be early events of breast tumorigenesis.
Several oncogenes and tumor suppressors that act as Pol I co-activators or co-repressors are known to modulate rRNA synthesis.Citation1,2 We found evidence that either overexpression of the MYC oncogene, a well-known rDNA activator,Citation8,9 or knockdown of the tumor suppressor MTG16a, an rDNA repressor,Citation4 in a human mammary epithelial cell context (HME1) lead to both rRNA synthesis upregulation and acquisition of a transformed phenotype.Citation4 Normal and transformed mammary epithelial cells can be distinguished based on the 3D morphology of acinar structures developed by cells when they are grown in a specific in vitro microenvironment.Citation10 By using this strategy, we found that both MYC-overexpressing and MTG16a knockdown HME1 cells, showing enlarged AgNORs as well as increased rRNA synthesis, develop into morphologically aberrant 3D acinar structures lacking the lumen, which is filled with proliferating cells, resembling the morphology of hyper-proliferative in situ breast cancer lesions.Citation4 In the present study we asked whether deregulation of basal components of the Pol I transcription machinery can per se impair mammary epithelial cell morphogenesis and contribute to breast cancer initiation, by inducing upregulation of rRNA synthesis.
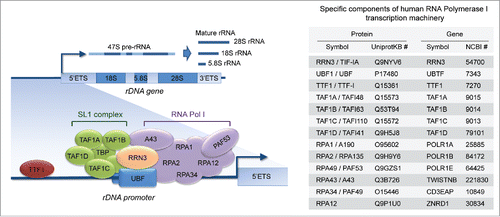
The assembly of the Pol I transcription machinery onto the rDNA promoter and the initiation of rDNA transcription require a large number of components (), many of which are specific to the Pol I transcription machinery (listed in ). The basal components of this machinery include, in addition to the subunits of RNA Polymerase I, many other proteins required for Pol I recruitment and activation of rDNA transcription. Some of the relevant factors are: the transcription initiation factor RRN3 (also known as TIF-IA), which serves as a bridge between Pol I and the pre-initiation complex at the rDNA promoter; the upstream binding factor UBF, which recruits Pol I to the rDNA promoter; the promoter selectivity factor SL1 (consisting of the TATA-binding protein, or TBP, and TBP-associated factors, or TAFs), which confers specificity for the Pol I promoter; the transcription terminator factor TTF1, which is required for both transcription termination and re-initiation of pre-rRNA synthesis.Citation1,11 According to The Cancer Genome Atlas (TCGA) and other published data sets,Citation12-16 many genes encoding for basal components of the Pol I transcription machinery are frequently amplified/upregulated in early in situ breast lesions, such as atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS), as well as in invasive breast carcinoma.
Figure 1. The basal components of the RNA-polymerase I (Pol I) transcription machinery. The assembly of the Pol I transcription machinery onto the ribosomal gene (rDNA) promoter and the initiation of rDNA transcription into pre-rRNA (subsequently processed into mature rRNAs) require several transcription factors and auxiliary proteins (left), many of which are specific to Pol I (right).
Here we specifically focus on RRN3, a key Pol I basal component that plays a major role in modulating rDNA transcription in response to external stimuli, such as nutrients and cellular stress.Citation1 Due to the key role of RRN3, its expression and function are regulated with different modalities (e.g. transcriptionally by MYC,Citation17 post-translationally by ERK-dependent phosphorylation,Citation18 and by AKT-induced stabilization and translocation to the nucleolusCitation19). There is evidence that RRN3 loss-of-function in different mammalian cell contexts leads to decreased rRNA synthesis, inhibition of cell proliferation, and induction of cell differentiation,Citation18,20,21 while RRN3 gain-of-function promotes rRNA synthesis and fosters cell proliferation.Citation18,22 Thus, we reasoned that upregulation of RRN3 in a mammary epithelial cell is likely to have significant biological repercussions, which may contribute to breast cancer initiation and progression.
Mechanistic studies shown here indicate that RRN3 knock down in the MCF7 breast cancer cell context significantly inhibits rRNA synthesis and cell proliferation, while ectopic expression of RRN3 in the HME1 cell context, by inducing rRNA upregulation, confers the potential for developing into morphologically aberrant 3D acinar structures with DCIS-like features. Moreover, we show that RRN3-induced rRNA upregulation sensitizes HME1 cells to the anti-proliferative action of a new generation Pol I inhibitor drug. Taken together these findings suggest that upregulation of RRN3, an essential basal component of the Pol I transcription machinery, by increasing rRNA synthesis, not only contributes to drive breast cancer cell proliferation, but can also be sufficient to trigger breast cancer initiation by compromising mammary epithelial morphogenetic processes.
Results
Amplification/upregulation of basal components of the Pol I transcription machinery in invasive breast cancer
The assembly of the Pol I transcription machinery onto the rDNA promoter and the initiation of rDNA transcription require a number of basal transcription factors and auxiliary proteins. We set out to analyze the mutational status and expression of genes encoding for 13 specific components of the Pol I transcription machinery () in 1105 cases of invasive breast carcinoma in the TCGA database. As shown in detail in and summarized in , about 66% of invasive breast carcinoma cases show alterations in one or more of these genes. Remarkably, more than 90% of altered genes were amplified and/or upregulated. In contrast, there were only a few missense mutations, and none of these mutations was recurrent or predicted to have high functional impact by Mutation Assessor analysis (http://mutationassessor.org/) (data not shown). Among the genes showing the highest percentage of amplification/upregulation we found TAF1A (26%) RRN3 (16%), ZNRD1 (9%) and CD3EAP (9%) (). Moreover, we detected concomitant amplification/upregulation of 2 or more genes in almost 50% of the cases (). Finally, we found that patients with amplification/upregulation of at least one gene encoding a basal component of the Pol I transcription machinery had a significantly (p = 0.014) worse prognosis than patients without alterations of these genes (median survival = 103 vs. 144 months) (). When we took into consideration cases with amplification/upregulation of at least 2 genes the prognosis difference became even more significant (p = 0.003) ().
Figure 2. Amplification/upregulation of basal components of the Pol I transcription machinery in invasive breast cancer. (A) TCGA analysis shows that genes encoding for basal components of the Pol I transcription machinery are frequently altered in invasive breast carcinoma. (B) Most of the altered genes are amplified and/or upregulated (left); almost half of these cases show concomitant amplification/upregulation of 2 or more genes (right). (C) Patients with amplification/upregulation of at least 1 (left) or at least 2 (right) genes encoding for basal components of the Pol I machinery have a significantly worse prognosis than patients with no alterations in these genes.
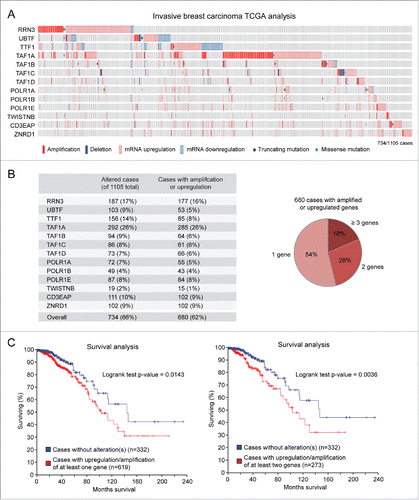
Taken together, these findings indicated that amplification/upregulation of one or more genes encoding basal components of the Pol I transcription machinery is frequent in invasive breast cancer, and is associated with a poor prognosis.
Detection of genomic gain of basal components of the Pol I transcription machinery in early breast cancer lesions
Enlarged AgNORs provide an indirect evidence of rRNA upregulation not only in invasive ductal carcinoma (IDC), but also in premalignant breast lesions and in situ breast carcinoma (e.g., DCIS).Citation6,7 Thus, we next searched for alterations of genes encoding for components of the Pol I transcription machinery in early breast cancer lesions. To this end, we first analyzed a published data set from a meta-analysis of 26 studies reporting copy number alterations (CNAs) in a total of 288 cases of “pure” DCIS (i.e. DCIS with no signs of invasion in the adjacent tissue) and 50 cases of atypical ductal hyperplasia (ADH).Citation16 As shown in , both DCIS and ADH showed genomic gain of regions containing genes of the Pol I transcription machinery. Remarkably, the genes amplified in the highest percentage of ADH and DCIS cases (e.g. RRN3 and TAF1A) were also amplified/upregulated in the TCGA dataset of breast invasive carcinoma (see ), suggesting that amplification/upregulation of Pol I components at more advanced breast cancer stages likely originated already at early stages. Indeed, when we analyzed published data sets from 3 independent studies that compared the genomic profiles of matched DCIS and IDC by comparative genomic hybridization (CGH) analysis of microdissected tissue,Citation13-15 we found that 23 out of 32 DCIS/IDC pairs showed genomic gain of the same regions containing genes of the Pol I transcription machinery ().
Figure 3. Detection of genomic gain of basal components of the Pol I transcription machinery in early breast cancer lesions. (A-B) Analysis of published data setsCitation13-16 shows that genes encoding for basal components of the Pol I transcription machinery are frequently amplified in ADH and pure DCIS (A) as well as in matched DCIS/IDC pairs (B).
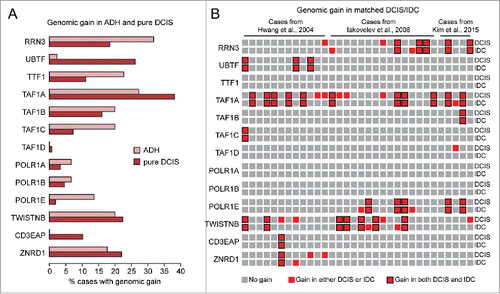
Overall, these analyses showed that upregulation of basal components of the Pol I transcription machinery is frequent not only in invasive breast cancer, but also at early stages of breast tumorigenesis, thus supporting the hypothesis that enhanced rRNA synthesis due to upregulation of these components can contribute to both breast cancer initiation and progression. To start tackling this hypothesis, we specifically focused on RRN3 because a) RRN3 is one of the Pol I components most prominently amplified/upregulated in both early and invasive breast cancer lesions, and b) it was previously reported that RRN3 expression and activity are critically linked to rRNA synthesis, cell differentiation, and cell proliferation in other cell models.Citation18,20-23
RRN3 knock down in MCF7 breast cancer cells leads to decreased rRNA transcription and inhibition of cell proliferation
We first tested whether RRN3-induced upregulation of rRNA synthesis contributes to breast cancer cell proliferation. To identify a suitable breast cancer cell model to perform loss-of-function experiments, we initially screened a panel of breast cancer cell lines (Hs-578T, MCF7, T47D, MDA-MB-468, and MDA-MB-231) for both RRN3 mRNA level and rRNA transcription. The latter was evaluated by measuring the level of pre-rRNA by qRT-PCR with primers amplifying a region in the rDNA 5′ETS, which is removed from mature rRNA (see scheme in , left). RRN3 was significantly higher (albeit at different degrees) in most breast cancer cell lines relative to non-tumorigenic HME1 mammary epithelial cells (). Consistently, cell lines with the highest RRN3 level (MCF7 and T47D) also displayed the highest level of pre-rRNA (), suggesting a causal link between upregulation of RRN3 and rRNA transcription in breast cancer cells.
Figure 4. RRN3 knock down in MCF7 breast cancer cells leads to decreased rRNA transcription and inhibition of cell proliferation. (A) RRN3 mRNA level (left) is significantly higher in breast cancer cell lines (yellow) relative to HME1 mammary epithelial cells (gray), and correlates with increased rRNA transcription (assessed as pre-rRNA level) (right). (B) Transient transfection of MCF7 with RRN3 siRNA (10 nM, 48h) decreases RRN3 mRNA (left, top), RRN3 protein (left, bottom), and RRN3 nucleolar accumulation (right) relative to cells transfected with a negative control scrambled sequence (siCtrl). (C-D) RRN3 knock down significantly reduces both rRNA synthesis (assessed by pre-rRNA qRT-PCR) (C) and cell proliferation (assessed by EdU incorporation) (D). * = p < 0.05; ** = p < 0.01; *** p < 0.001.
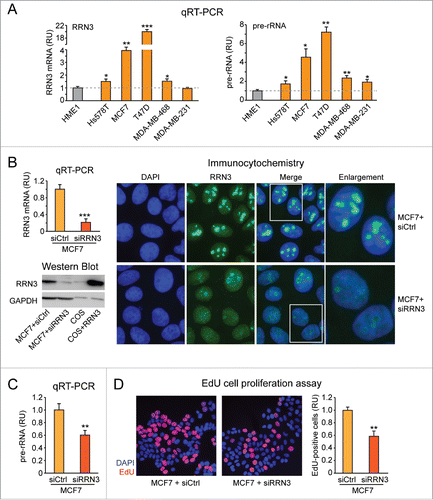
Next, we transiently knocked down RRN3 in MCF7, one of the 2 cell lines with the highest upregulation of both RRN3 and pre-rRNA. MCF7 cells transfected with RRN3 siRNA showed a significant reduction of both RRN3 mRNA () and RRN3 protein () relative to cells transfected with a scrambled negative control sequence (siCtrl). RRN3 knock down was also apparent by immunocytochemistry analysis, which showed reduced RRN3 accumulation in the nucleoli of MCF7 cells transfected with siRRN3 (). RRN3 knock down in MCF7 cells resulted in inhibition of both rRNA synthesis (see reduced pre-rRNA level in ) and cell proliferation (see reduced number of EdU-positive cells in ).
Based on these findings, RRN3 upregulation in MCF7 breast cancer cells apparently contributes to sustain the increased rate of both rRNA synthesis and proliferation. Since RRN3 is frequently amplified in early breast cancer lesions (see ), we next asked whether upregulation of RRN3 in a non-tumorigenic mammary epithelial cell context, by enhancing rRNA synthesis, is capable of inducing transformation.
Ectopic expression of RRN3 in HME1 cells promotes rRNA synthesis and confers increased sensitivity to the anti-proliferative action of the Pol I inhibitor CX-5461
To test if RRN3 overexpression is sufficient to induce upregulation of rRNA synthesis in human mammary epithelial cells, we developed stable HME1 clones overexpressing RRN3 (HME1-RRN3). As shown by quantitative western blot analysis, HME1-RRN3 cells expressed RRN3 protein at a significantly higher level relative to control HME1 cells (HME1-Ctrl), but similar to MCF7 breast cancer cells (a representative HME1-RRN3 clone is shown in ). The higher RRN3 level in HME1-RRN3 and MCF7 cells was also paralleled by a greater accumulation of RRN3 in the nucleolus (). Remarkably, HME1-RRN3 cells displayed significant rRNA upregulation relative to HME1-Ctrl cells (2 independent HME1-RRN3 clones are shown in ).
Figure 5. Ectopic expression of RRN3 in HME1 cells promotes rRNA synthesis and confers increased sensitivity to the anti-proliferative action of the Pol I inhibitor CX-5461. (A) Quantitative Western blot (blot shown at top left, quantification shown at bottom left) and immunocytochemistry (right) show that RRN3 protein expression in HME1 cells stably transfected with RRN3 (here shown a representative clone, HME1-RRN3-1) is higher relative to control HME1 cells transfected with the cognate empty vector (HME1-Ctrl), but similar to the RRN3 protein expression level of MCF7 breast cancer cells. (B) RRN3 overexpression leads to increased rRNA synthesis (assessed by pre-rRNA qRT-PCR) in 2 HME1-RRN3 clones relative to HME1-Ctrl cells. (C) MTT assay shows that the Pol I inhibitor CX-5461, which selectively inhibits Pol I transcription by disrupting the formation of the Pol I pre-initiation complex (left), inhibits proliferation of HME1-RRN3 cells at lower concentrations relative to HME1-Ctrl cells (right). (D) EdU incorporation assay further shows that 10 nM CX-5461 reduces the number of HME1-RRN3 cells in S phase (EdU-positive cells) while it does not affect HME1-Ctrl cells. * = p < 0.05; ** = p < 0.01.
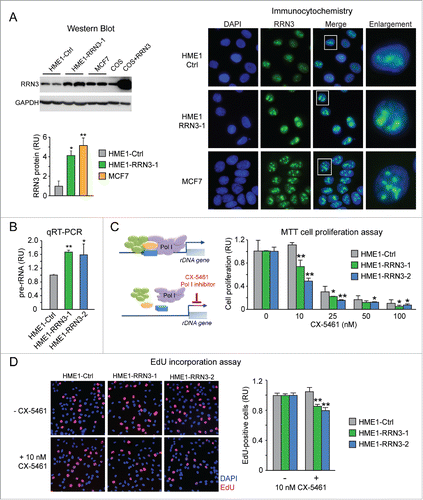
We next tested whether RRN3-induced rRNA upregulation affected the response to CX-5461, a new generation Pol I inhibitor known to selectively inhibit proliferation of cancer cells with rRNA upregulation by preventing the formation of the Pol I pre-initiation complex ().Citation24 As shown by MTT assay in , CX-5461 significantly inhibited HME1-RRN3 cell proliferation at lower concentrations relative to HME1-Ctrl cells. In particular, treatment with 10 nM CX-5461 significantly reduced proliferation of HME1-RRN3 cells, while it did not affect proliferation of HME1-Ctrl cells. These results were confirmed by EdU incorporation assay, showing that 10 nM CX-5461 significantly reduced the number of cells in S phase (EdU-positive cells) in HME1-RRN3, but not in HME1-Ctrl ().
Altogether these findings indicate that overexpression of RRN3 in HME1 mammary epithelial cells is sufficient to induce rRNA upregulation, and that inhibition of rRNA synthesis by a selective Pol I inhibitor exerts a significant anti-proliferative effect.
Stable RRN3-induced rRNA upregulation impairs 3D HME1 mammary epithelial morphogenesis
As we mentioned before, in vitro 3D mammary epithelial cell systems, including the HME1 mammary epithelial cell system used in this study, can effectively discriminate between normal and transformed mammary epithelial cells based on the 3D morphogenetic features acquired by cells in response to developmental cues when grown in a microenvironment.Citation4,10 To test whether RRN3-induced rRNA upregulation affected the morphogenetic potential of HME1 cells, we seeded HME1-Ctrl and HME1-RRN3 cells on reconstituted basement membrane (Matrigel) and assessed, after 10–12 days, the 3D acinar morphology by confocal microscopy. As shown in , HME1-Ctrl cells mostly developed into mature 3D acini with a lumen lined by polarized, growth-arrested cells. At this mature stage, Golgi and integrin staining clearly identified apico-basal and baso-lateral polarity within most acini, and EdU incorporation did not detect any actively dividing cell (). In contrast, HME1-RRN3 clonal lines mostly developed into aberrant 3D acini characterized by partially, or completely, filled lumen and presence of EdU-positive proliferating cells ().
Figure 6. Stable RRN3-induced rRNA upregulation impairs 3D HME1 mammary epithelial morphogenesis. Confocal analysis shows that HME1-Ctrl cells develop into morphologically normal acinar structures in 3-dimensional (3D) culture (left), while HME1-RRN3-1 and HME1-RRN3-2 clones, overexpressing RRN3, mostly form amorphous 3D acini characterized by partially, or completely, filled lumen and presence of proliferating cells (right). DAPI (blue) identifies nuclei, while Golgi apparatus (red)/integrin (green) staining detects cell polarity. Nuclei of proliferating cells were identified by EdU staining (red).
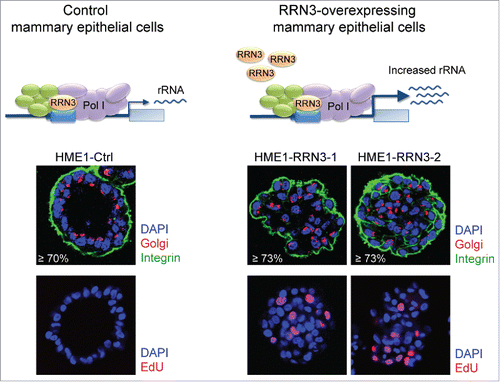
These findings show that upregulation of RRN3, by increasing rRNA synthesis, is sufficient to hamper HME1 3D mammary epithelial morphogenesis.
Basal components of the Pol I transcription machinery are amplified/upregulated in cancers of different histotype
Increased rRNA synthesis and ribogenesis, detected as enlarged AgNORs, have been reported in many cancers.Citation25 Indeed, when we analyzed the TCGA database for genes encoding basal components of the Pol I transcription machinery (see ), we detected alterations of these genes in tumors of different histotype (). The overall percentage of gene alterations ranged from 18% to over 65%. When single genes were taken into account, the percentage of altered cases varied among different cancers. For instance, RRN3 showed the highest percentage of mutation/deregulation in breast cancer (17%), TAF1A in breast (26%) and liver cancer (17%), TWISTNB in lung adenocarcinoma (31%) and testicular cancer (31%), ZNRD1 in skin melanoma (18%), TTF1 in cervical carcinoma (18%). In all cancers the majority of the genes analyzed was mainly upregulated and/or amplified (see red portion of the pie charts in ). In some cancers (e.g., stomach adenocarcinoma) and for some genes (e.g. POLR1A), we also detected several missense mutations, but Mutation Assessor analysis did not identify any recurrent mutation with high functional impact (data not shown).
Figure 7. Basal components of the Pol I transcription machinery are amplified/upregulated in cancers of different histotype. TCGA analysis shows that genes encoding for basal components of the Pol I transcription machinery are frequently amplified/upregulated in different cancer types (see Results and Methods for details).
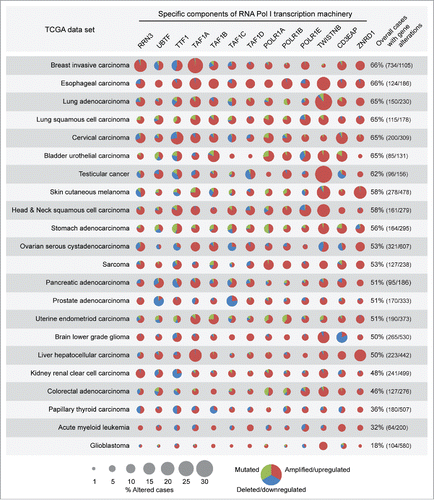
Apparently, amplification/upregulation of genes encoding basal components of the Pol I transcription machinery is detectable in most cancers. It remains to be determined whether upregulation of rRNA synthesis is sufficient to initiate transformation of cells of different histotype.
Discussion
For decades pathologists have recognized that an enlarged nucleolar compartment, which mirrors increased rRNA transcription and ribogenesis, is a hallmark of cancer.Citation5,26 Both overexpression of oncogenes that act as Pol I activators and loss of tumor suppressors that act as Pol I inhibitors were reported to enhance rRNA synthesis in cancer cells.Citation1,2 However, there is evidence that rRNA deregulation in cancer can be caused by other factors, including epigenetic mechanisms,Citation27 given that rRNA transcription is epigenetically regulated at the chromatin level.Citation28
In this study we provide evidence that upregulation of basal components of the Pol I transcription machinery itself can per se lead to increased rRNA synthesis in cancer cells. First, the TCGA let us detect several genes encoding basal components of the Pol I transcription machinery that are very frequently (66%) amplified/upregulated in invasive breast cancer. Notably, in almost half of these cases 2 or more genes of the Pol I transcription machinery are amplified/upregulated, making us speculate that the concomitant upregulation of these genes may contribute to maintain the correct stoichiometry of the components of the Pol I transcription machinery. Furthermore, from other published datasetsCitation13-16 we detected amplification of genes of the Pol I transcription machinery in ADH and DCIS, consistent with previous studies reporting the detection of enlarged AgNORs in both premalignant and early breast disease stages.Citation6,7 Amplification of these genes was detected also in DCIS and matched adjacent IDC tissue, suggesting that upregulation of basal components of the Pol I transcription machinery plays a role in both breast cancer initiation and progression. Here we focused on one of these basal components, RRN3, because studies in different cell contexts show that its overexpression/constitutive activation induces rRNA synthesis and drives cell proliferation, while delaying cell differentiation.Citation18,22,23 Moreover, as shown here, RRN3 is frequently amplified/upregulated in both early breast cancer lesions and invasive breast cancer, and its upregulation does correlate with pre-rRNA upregulation in breast cancer cell lines. Consistently, RRN3-overexpression in the HME1 mammary epithelial cell context, by increasing rRNA synthesis, hampers normal 3D morphogenesis, thus leading to the formation of DCIS-like acinar structures. Conversely, knock down of RRN3 in the MCF7 breast cancer cell context is sufficient to significantly reduce pre-RNA level and inhibit cell proliferation. These complementary findings suggest that increased rRNA synthesis due to RRN3 upregulation, by impairing mammary epithelial cell morphogenetic processes and by sustaining cancer cell proliferation, contributes to breast tumorigenesis.
In this study, we also show that RRN3-induced rRNA upregulation in breast cancer might become a promising therapeutic target, based on the observation that RRN3-induced rRNA synthesis makes HME1 cells more sensitive to the anti-proliferative effects of CX-5461, a new generation Pol I inhibitor. CX-5461 has proved to be an effective cancer agent that selectively targets increased rRNA transcription in cancer cells while sparing normal cells.Citation5,24,29-31 CX-5461 is currently being tested in breast cancer clinical trials.Citation32 In addition to CX-5461, there are also new molecules targeting RRN3 directly, such as a recently described small molecule that inhibits the interaction between Rrn3 and Pol I in yeast.Citation33 This molecule needs to be further developed because it may be effective at selectively targeting RRN3-overexpressing breast cancer.
In summary, this study suggests that increased rRNA synthesis due to RRN3 upregulation, by affecting mammary epithelial morphogenetic processes and sustaining breast cancer cell proliferation, can contribute to breast cancer initiation and progression, and might be targeted with new small molecules. This study is also relevant since upregulation of rRNA synthesis due to amplification/upregulation of RRN3 and other genes encoding basal components of the Pol I transcription machinery, which is frequent in many different cancers (), might be an etiological factor more common than previously expected.
Material and methods
TCGA analysis
Analysis of The Cancer Genome Atlas (TCGA) data sets was performed through the cBioPortal for Cancer Genomics (http://www.cbioportal.org/public-portal/).Citation34,35 Analysis was performed on all available cases (“all tumors” option) of the following TCGA datasets as of February 2016: breast invasive carcinoma, esophageal carcinoma, lung adenocarcinoma (Nature 2014), lung squamous cell carcinoma (Nature 2012), cervical squamous cell carcinoma and endocervical adenocarcinoma, testicular germ cell cancer, bladder urothelial carcinoma (Nature 2014), skin cutaneous melanoma, head and neck squamous cell carcinoma (Nature 2015), ovarian serous cystadenocarcinoma, stomach adenocarcinoma (Nature 2014), pancreatic adenocarcinoma, sarcoma, prostate adenocarcinoma (Cell 2015), brain lower grade glioma, liver hepatocellular carcinoma, uterine corpus endometrioid carcinoma (Nature 2013), kidney renal clear cell carcinoma (Nature 2013), colorectal adenocarcinoma (Nature 2012), papillary thyroid carcinoma (Cell 2014), acute myeloid leukemia (NEJM 2013), glioblastoma (Cell 2013). For each TCGA cancer data set, the following user-defined gene set was analyzed: RRN3, UBTF, TTF1, TAF1A, TAF1B, TAF1C, TAF1D, POLR1A, POLR1B, POLR1E, TWISTNB, CD3EAP, ZNRD1. The following genomic profile options were selected for each analysis: mutations, putative copy-number alterations from GISTIC, mRNA expression data (mRNA Expression z-Scores, RNA Seq V2 RSEM, z-score threshold ± 2.0). TCGA analysis results were exported and manually analyzed to calculate frequencies of genes amplified/upregulated, deleted/downregulated, or mutated. Survival analyses were performed in cBioPortal by uploading user-defined case lists comprising only cases with amplification/upregulation and cases with no alterations of the above-mentioned genes. Mutation analysis was performed by using the Mutation Assessor tool (http://mutationassessor.org/) in cBioPortal.
Analysis of ADH and DCIS datasets
A published data set from a meta-analysis of 26 studiesCitation16 was used to assess genomic gains in pure DCIS and ADH, while datasets from 3 independent studiesCitation13-15 were used to assess genomic gains in matched DCIS and IDC. Genomic gain of the genes encoding basal components of the Pol I transcription machinery was manually determined based on gain of the corresponding cytoband. The cytobands corresponding to each gene were obtained by comparing the gene location on the chromosome with the cytoband genomic location indicated in the UCSC cytoband table.
Cells and cell culture
COS cells and the MCF7, T47D, Hs578T, MDA-MB-468, and MDA-MB-231 breast cancer cell lines (ATCC) were grown as per ATCC recommendations in a humidified incubator with 5% CO2 at 37 °C. The telomerase-immortalized human mammary epithelial cell line h-TERT-HME1 (here referred to as HME1) (Clontech) and derived clones were grown in MEGM (Lonza).
For transient RRN3 knock down in MCF7, 105 cells/well were seeded in 24 well plates on coverslips, let attach overnight, and transfected with either 10 nM siRRN3 (Silencer Select pre-designed siRNA, ID: s29324, Thermo Fisher) or 10 nM siRNA Negative Control (Thermo Fisher) by using RNAi max Lipofectamine (Thermo Fisher) according to the manufacturer's instructions. 48 h after transfection RRN3 knock down was assessed by qRT-PCR, Western blot and immunocytochemistry.
To develop clones stably expressing ectopic RRN3, HME1 cells were transfected with pcDNA3.1-RRN3 (kindly provided by Dr. Grummt's lab, Heidelberg) by using Lipofectamine LTX (Thermo Fisher). Control clones were developed by stable transfection with empty pcDNA3.1. After selection with 1 mg/ml G418, single clones were isolated with cloning rings, expanded and analyzed for the presence of the transfected construct by PCR as well as for RRN3 expression by western blot and immunocytochemistry.
For treatments with CX-5461 (ApexBio), the drug was dissolved in 50 mM NaH2PO4 (pH 4.5) to obtain a 1 mM stock solution and stored at −20°C. After seeding, cells were let attach overnight and treated with the indicated concentrations of CX-5461 in growth medium. Medium was refreshed every 24 hours for the duration of the treatment.
3D culture was performed as we described.Citation4 Briefly, cells were seeded in 8 well chamber slides at low density (3 × 103 single cells/well) on a layer of growth-factor reduced Matrigel (BD Biosciences) covered with growth medium plus 2% Matrigel. Cells were let grow for 10–12 d until they formed mature acini, refreshing the medium every 2–3 d.
Western blot
Equal numbers of cells were lysed in Laemmli buffer to obtain full lysis of all nuclear/nucleolar components. COS cells transiently transfected with or without pcDNA3.1-RRN3 (transfections performed by using Lipofectamine LTX, Thermo Fisher, according to the manufacturer's instructions) were used as a control for RRN3 expression. Protein lysates were separated by SDS PAGE electrophoresis and blotted onto a PVDF membrane according to standard protocols. After transfer, membranes were blocked with 5% non-fat dry milk in TBS + 0.05% Tween 20 for 1 h at room temperature, incubated with anti-RRN3 antibody (HPA049837, Atlas Antibodies) diluted in 5% milk over night at 4 °C, washed, incubated with HRP-conjugated anti-rabbit antibody (GE Life Sciences) diluted in 5% milk for 1 h at room temperature, washed, and reacted with Clarity ECL (Biorad). Chemiluminescent signal was detected and quantified by using ChemiDoc Touch Imaging System (Biorad). Statistical significance was calculated by using the Student's t-test.
Immunocytochemistry
For RRN3 immunostaining in standard (2D) culture, subconfluent cells grown on coverslips in 24 well plates were fixed with 4% paraformaldehyde for 10 min, permeabilized with PBS+0.1% Triton X100 for 15 min, blocked with PBS + 4% PBS for 20 min, incubated with anti-RRN3 (Atlas Antibodies) diluted in PBS+4% FBS over night at 4 °C, washed 4 times with PBS, incubated with anti-rabbit AlexaFluor 488 (Thermo Fisher) diluted in PBS + 4% FBS for 1 hour, washed 4 times with PBS, counterstained with DAPI, and mounted with Vectashield (Vector Laboratories). Images were analyzed at a fluorescence microscope. The same exposure time was used for all samples.
For immunostaining of 3D acini, immunocytochemistry was performed as we previously described.Citation4 Briefly, mature 3D acini were fixed with 4% paraformaldehyde for 15 min., permeabilized with PBS + 0.2% Triton X100 for 10 min., blocked with PBS + 1% BSA, 1% FBS and 0.05% Tween 20 for 1 hour, incubated with the primary antibody overnight at 4 °C, rinsed, and incubated with the appropriate secondary antibody for 2 h at room temperature, rinsed, counterstained with DAPI (Sigma), and mounted with Vectashield (Vector Laboratories). The Golgi apparatus was detected with anti-GM130 antibody (BD Biosciences) and integrin was stained with anti-CD49f antibody (EMD Millipore, Billerica, MA). Acini were analyzed by confocal microscopy (SP2 Spectral Confocal Microscope, Leica) and classified as normal or aberrant based on their morphology.
qRT-PCR
Total RNA was extracted with Trizol (Thermo Fisher), treated with DNase I (Thermo Fisher), and retrotranscribed with random primers using High Capacity cDNA RT kit (Thermo Fisher). cDNA was amplified by real time PCR by using the iQ SYBR Green Supermix (Bio-Rad) with primers specific for human pre-rRNA 5′ETS (sense: 5′-CTGAGGGAGCTCGTCGGTGT-3′, antisense: 5′-GCAGAGCGCCTCCGAAGTCA-3′), RRN3 (sense: 5′-CGGAAACCTGAAAGAAGGTTTGC-3′, antisense: 5′-CTGGCGATTGTTCCTCTCAATG-3′), or GAPDH (sense: 5′-GAAGGTGAAGGTCGGAGTC-3′, antisense: 5′-GAAGATGGTGATGGGATTTC-3′) as we previously described.Citation4 Statistical significance was calculated by using the Student's t-test.
MTT assay
2 × 103 cells/well were seeded in 24 well plates in 4 replicates, let attach overnight, and treated with CX-5461. After one week of treatment, cells were incubated with 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) for 2 hours under standard growth conditions and lysed with DMSO. Cell lysate absorbance at 570 nm was measured on a microplate spectrophotometer. Statistical significance was calculated by using the Student's t-test.
EdU incorporation assay
The number of cells in S phase was assessed by measuring EdU-incorporation with Click-iT EdU imaging kit (Thermo Fisher), according to the manufacturer's protocol. Prior to fixation cells were incubated under standard growth conditions with either 10 µM EdU for 1 h (2D culture) or 40 µM EdU for 2 h (3D culture). After incorporation, cells were fixed with 4% paraformaldehyde for 20 min., permeabilized with PBS + 0.5 % Triton X100 for 20 min., washed with PBS + 3% BSA, incubated with Click-iT reaction cocktail (with Alexa Fluor 594) for 30 min., counterstained with DAPI, and mounted with Vectashield (Vector Laboratories). EdU-positive and DAPI-positive cells were counted in at least 10 random fields. Statistical significance was calculated by using the Student's t-test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Ingrid Grummt, German Cancer Research Center, Heidelberg, and Raffaella Santoro, University of Zurich, for providing us with the pcDNA3.1-RRN3(TIF-IA) construct and for helpful discussions.
Funding
Funding for this study was provided by the Terri Brodeur Breast Cancer Foundation (SR), the Susan Komen Foundation (SR), the NCI R01 CA127614 grant (NS), and the NCI P30 CA016056 institutional grant.
Related Research Data
References
- Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol 2010; 50:131-56; PMID:20055700; http://dx.doi.org/10.1146/annurev.pharmtox.010909.105844
- Nguyen le XT, Raval A, Garcia JS, Mitchell BS. Regulation of ribosomal gene expression in cancer. J Cell Physiol 2015; 230:1181-8; PMID:25336383; http://dx.doi.org/10.1002/jcp.24854
- Hannan KM, Sanij E, Rothblum LI, Hannan RD, Pearson RB. Dysregulation of RNA polymerase I transcription during disease. Biochim Biophys Acta 2013; 1829:342-60; PMID:23153826; http://dx.doi.org/10.1016/j.bbagrm.2012.10.014
- Rossetti S, Hoogeveen AT, Esposito J, Sacchi N. Loss of MTG16a (CBFA2T3), a novel rDNA repressor, leads to increased ribogenesis and disruption of breast acinar morphogenesis. J Cell Mol Med 2010; 14:1358-70; PMID:19961547; http://dx.doi.org/10.1111/j.1582-4934.2009.00982.x
- Ruggero D. Revisiting the nucleolus: from marker to dynamic integrator of cancer signaling. Sci Signal 2012; 5:pe38; PMID:22969157; http://dx.doi.org/10.1126/scisignal.2003477
- Ceccarelli C, Trere D, Santini D, Taffurelli M, Chieco P, Derenzini M. AgNORs in breast tumours. Micron 2000; 31:143-9; PMID:10588060; http://dx.doi.org/10.1016/S0968-4328(99)00071-2
- Derenzini M, Betts CM, Trere D, Mambelli V, Millis RR, Eusebi V, Cancellieri A. Diagnostic value of silver-stained interphasic nucleolar organizer regions in breast tumors. Ultrastruct Pathol 1990; 14:233-45; PMID:1694051; http://dx.doi.org/10.3109/01913129009076127
- Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol 2005; 7:303-10; PMID:15723053; http://dx.doi.org/10.1038/ncb1225
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 2005; 7:311-8; PMID:15723054; http://dx.doi.org/10.1038/ncb1224
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A 1992; 89:9064-8; PMID:1384042; http://dx.doi.org/10.1073/pnas.89.19.9064
- Goodfellow SJ, Zomerdijk JC. Basic mechanisms in RNA polymerase I transcription of the ribosomal RNA genes. Sub-Cell Biochem 2013; 61:211-36; PMID:23150253; http://dx.doi.org/10.1007/978-94-007-4525-4_10
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015; 163:506-19; PMID:26451490; http://dx.doi.org/10.1016/j.cell.2015.09.033
- Hwang ES, DeVries S, Chew KL, Moore DH, 2nd, Kerlikowske K, Thor A, Ljung BM, Waldman FM. Patterns of chromosomal alterations in breast ductal carcinoma in situ. Clin Cancer Res 2004; 10:5160-7; PMID:15297420; http://dx.doi.org/10.1158/1078-0432.CCR-04-0165
- Iakovlev VV, Arneson NC, Wong V, Wang C, Leung S, Iakovleva G, Warren K, Pintilie M, Done SJ. Genomic differences between pure ductal carcinoma in situ of the breast and that associated with invasive disease: a calibrated aCGH study. Clin Cancer Res 2008; 14:4446-54; PMID:18628458; http://dx.doi.org/10.1158/1078-0432.CCR-07-4960
- Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget 2015; 6:7597-607; PMID:25831047; http://dx.doi.org/10.18632/oncotarget.3162
- Rane SU, Mirza H, Grigoriadis A, Pinder SE. Selection and evolution in the genomic landscape of copy number alterations in ductal carcinoma in situ (DCIS) and its progression to invasive carcinoma of ductal/no special type: a meta-analysis. Breast Cancer Res Treat 2015; 153:101-21; PMID:26255059; http://dx.doi.org/10.1007/s10549-015-3509-x
- Poortinga G, Wall M, Sanij E, Siwicki K, Ellul J, Brown D, Holloway TP, Hannan RD, McArthur GA. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res 2011; 39:3267-81; PMID:21177653; http://dx.doi.org/10.1093/nar/gkq1205
- Zhao J, Yuan X, Frodin M, Grummt I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell 2003; 11:405-13; PMID:12620228; http://dx.doi.org/10.1016/S1097-2765(03)00036-4
- Nguyen le XT, Mitchell BS. Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc Natl Acad Sci U S A 2013; 110:20681-6; PMID:24297901; http://dx.doi.org/10.1073/pnas.1313097110
- Hayashi Y, Kuroda T, Kishimoto H, Wang C, Iwama A, Kimura K. Downregulation of rRNA transcription triggers cell differentiation. PloS One 2014; 9:e98586; PMID:24879416; http://dx.doi.org/10.1371/journal.pone.0098586
- Yuan X, Zhou Y, Casanova E, Chai M, Kiss E, Grone HJ, Schutz G, Grummt I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell 2005; 19:77-87; PMID:15989966; http://dx.doi.org/10.1016/j.molcel.2005.05.023
- Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev 2004; 18:423-34; PMID:15004009; http://dx.doi.org/10.1101/gad.285504
- Zhang Q, Shalaby NA, Buszczak M. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 2014; 343:298-301; PMID:24436420; http://dx.doi.org/10.1126/science.1246384
- Drygin D, Lin A, Bliesath J, Ho CB, O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res 2011; 71:1418-30; PMID:21159662; http://dx.doi.org/10.1158/0008-5472.CAN-10-1728
- Derenzini M, Montanaro L, Trere D. What the nucleolus says to a tumour pathologist. Histopathology 2009; 54:753-62; PMID:19178588; http://dx.doi.org/10.1111/j.1365-2559.2008.03168.x
- Pianese G. Beitrag zur histologie und aetiologie der carcinoma. histologische und experimentelle untersuchungen. Beitr Pathol Anat Allgem Pathol 1896; 142:1-193
- Bacalini MG, Pacilli A, Giuliani C, Penzo M, Trere D, Pirazzini C, Salvioli S, Franceschi C, Montanaro L, Garagnani P. The nucleolar size is associated to the methylation status of ribosomal DNA in breast carcinomas. BMC Cancer 2014; 14:361; PMID:24884608; http://dx.doi.org/10.1186/1471-2407-14-361
- Grummt I, Langst G. Epigenetic control of RNA polymerase I transcription in mammalian cells. Biochim Biophys Acta 2013; 1829:393-404; PMID:23063748; http://dx.doi.org/10.1016/j.bbagrm.2012.10.004
- Leslie M. Central command. Science 2014; 345:506-7; PMID:25082681; http://dx.doi.org/10.1126/science.345.6196.506
- Negi SS, Brown P. Transient rRNA synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 2015; 6:34846-58; PMID:26472108; http://dx.doi.org/10.18632/oncotarget.4093
- Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD. Targeting the nucleolus for cancer intervention. Biochim Biophys Acta 2014; 1842:802-16; PMID:24389329; http://dx.doi.org/10.1016/j.bbadis.2013.12.009
- Canadian Cancer Trials Groups. A Phase I/II Study of CX5461 (NCT02719977). ClinicalTrials.gov, 2016
- Rothblum K, Hu Q, Penrod Y, Rothblum LI. Selective inhibition of rDNA transcription by a small-molecule peptide that targets the interface between RNA polymerase I and Rrn3. Mol Cancer Res 2014; 12:1586-96; PMID:25033839; http://dx.doi.org/10.1158/1541-7786.MCR-14-0229
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/10.1158/2159-8290.CD-12-0095
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/10.1126/scisignal.2004088