
ABSTRACT
Inherited bone marrow failure syndromes (BMFS) are rare, distressing, inherited blood disorders of children. Although the genetic origin of these pathologies involves genes with different functions, all are associated with progressive haematopoietic impairment and an excessive risk of malignancies. Defects in energy metabolism induce oxidative stress, impaired energy production and an unbalanced ratio between ATP and AMP. This assumes an important role in self-renewal and differentiation in haematopoietic stem cells (HSC) and can play an important role in bone marrow failure. Defects in energetic/respiratory metabolism, in particular in FA and SDS cells, have been described recently and seem to be a pertinent argument in the discussion of the haematopoietic defect in BMFS, as an alternative to the hypotheses already established on this subject, which may shed new light on the evolution of these diseases.
The bone marrow failure syndromes (BMFS) include a group of disorders than can be either inherited or acquired, and share phenotypic hallmarks of age-related diseases. The aging process in particular affects haematopoietic stem cells, inducing reduced regenerative potential with a possible increase in genetically unstable cells and malignancy. Although the genetic origin of these pathologies involves genes with different functionsCitation1,2 (e.g., DNA repair, ribosome biogenesis, and telomere maintenance) there is a common final pathway, yet still largely elusive, by which HSC are unable to support the production of blood cells.Citation3
Telomere maintenance, DNA repair and oxidative stress are biochemically interrelated processes responsible for cellular aging. Even though the aging mechanism has not yet been clarified and several theories are proposed,Citation4 mitochondrial metabolism, reactive oxygen species (ROS) and cellular antioxidant systems maintain a central role in the aging process.Citation5,6 In fact, oxidative stress and ROS increment are a common factor in BMFS.Citation7
Fanconi's Anaemia (FA), Shwachman-Diamond Syndrome (SDS), Dyskeratosis Congenital (DC) and the Diamond-Blackfan Syndrome (DBA) are the most frequent inherited BMFS disorders. FA cells show chromosomal instability, hypersensitivity to crosslinking agents and defects of DNA repair mechanisms. These characteristics seem strikingly linked to defects in the oxidative stress response. In fact, chromosomal aberrations in FA lymphocytes are positively related to oxygen tension and spontaneous chromosomal instability is reduced by superoxide dismutase and catalase activity. Recently, it has been shown that antioxidant (AO) molecules improve haematopoiesis and reduce spontaneous and induced chromosomal instability in FA. Moreover, oxidative stress is a critical factor in the pathogenesis of bone marrow failure and leukemia progression in FA cells.Citation8,9
SDS cells have a defect in ribosome biogenesis, chemotaxis, mitotic spindle formation and cellular stress responses. Regarding the latter aspect, in SDS cells, endoplasmic reticulum and mitochondrial stress may enhance reactive oxygen species (ROS) production, leading to increased apoptosis and decreased cell growth.Citation10-12 DC is caused by mutations in genes involved in telomere maintenance, which induces sensitivity to DNA damage and increments of oxidative stress, sensitive to NAC treatment.Citation13,14
DBA depends on mutations in one of at least 10 ribosomal proteins necessary for the processing of pre-rRNA and the assembly of ribosomal subunits and show an increment of oxidative stress that regulates the autophagy process.Citation15 Also in this case, antioxidant treatment causes a reduction in autophagy in DBA cells.
Recently, we have demonstrated that FA and SDS are characterized by a decrease in energy production by the oxidative phosphorylation (OXPHOS) machinery.Citation16,17 These alterations cause a decrement in ATP production, determining an unbalanced ratio between ATP and AMP.Citation16 Whatever is the role of FA and SDS, it is important to note that the dysfunction of the OXPHOS machinery determines an increment in oxidative stress production, which induces lipid peroxidation and therefore, an alteration in the mitochondrial membrane, where the OXPHOS machinery resides.Citation16,17 This triggers a vicious cycle that increases the inefficiency of ATP and oxidative stress production.Citation18 To re-establish the correct energetic status, SDS and FA cells augment the glycolysis rate.Citation16,17 Hence, the return to glycolytic metabolism does not represent a Warburg effect, but the only choice of the cell to produce energy without increasing the accumulation of oxidative damage. However, it is also possible to speculate that this metabolic switch in a differentiated cell represents a precancerous condition that may facilitate tumor transformation. In DBA, the metabolic defects seem completely different. In fact, DBA cells show altered expression of several genes involved in energetic metabolism with reduced ATP levels and a switch from glycolytic to OXPHOS metabolism, with an augmentation of oxidative stress.Citation19 However, studies are needed to better understand energy metabolism in DBA cells.
Different topics can be discussed in relation to the energy problem in these cells. Mitochondrial activity is influenced by the integrity of the mitochondrial reticulum, due to the fusion-fission mechanism, and by their correlation with the endoplasmic reticulum, via mitochondria-associated membranes proteins (MAM).Citation20,21 In fact, frequent cycles of fusion and fission adapt the morphology of the mitochondrial compartment to the metabolic needs of the cell. Fusion is particularly important in metabolically active cells because it enables the distribution of metabolites, whereas mitochondrial fission plays an important role in the removal of damaged organelles by autophagy.Citation22 Thus, mitochondrial fusion and fission contribute to the maintenance of mitochondrial function and optimize bioenergetic capacity. In FA lymphocytes, mitochondria are swollen with matrix rarefaction and altered cristae, while in primary fibroblasts, fragmentation of the mitochondrial reticulum has been described.Citation16,23 Moreover, altered mitochondrial structures are also present in cell models of DBA, while human SDS lymphocytes show normal mitochondrial structure when observed under electron microscopy.Citation17,19 Moreover, the energetic function of mitochondria depends on different factors, including several signaling pathways. Among these, AMP-dependent protein kinase (AMPK) and the IP3K/AKT/mTOR pathway play a pivotal role. AMPK is an important sensor of cellular energy status activated by high concentrations of intracellular AMP.Citation24 AMPK activation stimulates alternative catabolic processes (such as glycolytic flux, glucose uptake, and fatty acid oxidation), generating ATP. At the same time, activated AMPK limits energy-consuming processes by antagonizing mTOR kinase, which is critical for translation initiation. In a simplified view, when energy is low, AMPK is active and mTOR is inhibited. mTOR signaling is necessary for the maintenance of mitochondrial oxidative functionCitation25 through a complex crosstalk, comprising Bcl-xl and VDAC1 at the mitochondrial outer membrane. It was observed that mTOR activates Bcl-xl, maintaining a sufficient ATP/ADP exchange between the mitochondria and cytosol to sustain coupled respiration, allowing the mitochondria to adapt to changes in metabolic demand.Citation26 In FA and SDS energetic defects of cells induce AMPK hyper-phosphorylation.Citation16,17,19,27 Surprisingly, the IP3K/AKT/mTOR pathway is also hyper-activated.Citation17,28,29 In DBA, hyper-activation of mTOR is congruent with the increment in OXPHOS over glycolytic metabolism, while in FA and SDS, hyper-activation of mTOR might suggest the cells' attempt to overcome the energy defect through glutaminolysis, a pathway used in particular by cancer cells to support their energy requirements. Moreover, hyper-activation of mTOR inhibits autophagy and these data fit with its reduction in FA,Citation30 even though in SDS and DBA, autophagy increases.Citation15
Finally, Ca2+ is a crucial signaling molecule in energy metabolism that regulates many cellular ATP-consuming reactions. Ca2+ signaling appears to be maintained through a finely tuned dynamic interplay between the endoplasmic reticulum, mitochondria and plasma membrane. Ca2+ uptake into mitochondria activates the tricarboxylic acid (TCA) cycle, which supplies NADH for oxidative phosphorylation. Electron transfer, coupled with proton pumping in the inter-mitochondrial membrane space, establishes the electrochemical potential used to convert ADP to ATP. Mitochondria, however, are also the most important source of free-radical production and have a crucial role in intracellular calcium concentration ([Ca2+]i) homeostasis and the consequent negative effects associated with the induction of intrinsic cell death.Citation31 Moreover, in response to energetic stress, increasing intracellular Ca2+ acts as a second messenger, leading to activation of AMPK.Citation32 Therefore, the alterations of the energetic status of FA and SDS cells could be related to an altered calcium level. In FA cells, the [Ca2+]i is dramatically low.Citation33 By contrast, SDS cells display a 2-fold elevation of [Ca2+]i relative to controls.Citation17 These data are very interesting, considering that Ca2+ has a dual effect on energetic metabolism: on the one hand, it enhances OXPHOS function, activating the Krebs' cycle and the mitochondrial substrate transportersCitation34; on the other hand, it inhibits complex IV, stimulating ROS production through the respiratory chain.Citation35 This implies that any alteration in mitochondrial [Ca2+]i impacts on aerobic energy metabolism,Citation31 but also limits oxidative stress production. Therefore, this mechanism, while leading to a decrement in ATP production, might allow, at the same time, an extension of the cell's life, delaying apoptosis events.Citation36
Biochemical defects in energetic/respiratory metabolism described in BMFS (represented in ) seem to be a pertinent argument to discuss the haematopoietic defect in these patients. Considering that no mutations related to the function or the organization of mitochondria have been described in the BMFS diseases treated in this work, it is possible to presume that functional mitochondrial impairment is actually a secondary defect with reference to the pathologic trait in the specific disease.
Figure 1. Schematic representation of a possible mechanism that induces bone marrow failure. ^DNA damage; *Oxidative stress; +Membrane peroxidation.
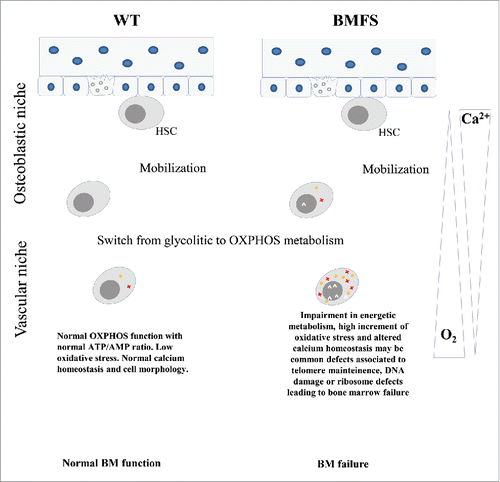
Stem cells are sensitive to increases in oxidative stress production. To counteract the effect of oxidative damage, stem cells are in a state of quiescence within a hypoxic microenvironment (niche) composed of different cells and molecular factors.Citation37 Stem cells possess 2 idiosyncratic characteristics: pluripotency, which allows mature cells to compose specific organs or tissues, and self-renewal, which supplies an organ with an adequate number of cells to maintain the organ's function. During self-renewing stem cell division, asymmetric segregation of mitochondria has been described. The daughter cells that retain pluripotent capacity inherit fewer mitochondria to protect the stem cells from increased oxidative risk, while the other ones increase in number and the activity of mitochondria and start the OXPHOS metabolism needed for the differentiation process.Citation38
In bone marrow, haematopoietic stem cells (HSC) are located in 2 different niches. The osteoblastic niche is closest to the endosteum with low oxygen concentration (hypoxia), which forces HSC to utilize glycolytic metabolism and protects the cells from oxidative stress induced by mitochondrial oxidative phosphorylation. Moving radially toward the longitudinal axis of the marrow into a more vascularised area, the vascular niche, HSC favor proliferation and differentiation, acquiring an oxido-reductive metabolic profile. The calcium ion gradient regulates the quiescent state of HSC by Ca2+ calmodulin (CaM)-dependent protein kinase IV.Citation39 Thus, the duplication of quiescent stem cells represents a delicate event, which must be strictly regulated. The passage from glycolytic to OXPHOS metabolism produces oxidative stress and DNA damage. Therefore, the damage caused by mitochondrial alteration becomes severe during differentiation or when metabolism switches from anaerobic to aerobic. The energetic metabolism assumes, therefore, an important role in self-renewal and differentiation. While under normal conditions, this switch represents a more efficient system to produce energy, its impairment represents a cause of the increment of oxidative stress products and of energetic defect, contributing to cellular damage and aging. Therefore, since HSC are subject to a continuous process of cell division and differentiation, BM represents a target organ in disease in which genetic alteration causes impairment in cellular respiratory and energy metabolism. Walter et al. recently described metabolic stress in HSC, observing that the induction of proliferation causes an increase in DNA damage induced by oxidative stress.Citation40
In particular, in HSC of FA mice, an augmentation of DNA damage was observed, along with cell death and reduced ability of stem cells to renew, underlining the DNA repair function of FA proteins in haematopoiesis. These results, however, underline the importance of DNA repair in FA, apart from the role of oxidative stress in this disease.
Different considerations could be made about the defect of energetic metabolism in DBA, where the glycolytic pathway appears altered, compromising the energetic source of HSC cells. However, the information concerning energy metabolism in these cells is yet incomplete and other studies are needed to better understand the biochemical metabolism of DBA cells, which, despite being a disease whose target is the erythroid lineage, upon differentiation will mandatorily imply glycolytic metabolism.
In conclusion, we can speculate that defects in energetic and respiratory metabolism play an important role in FA and SDS patients. If our assumption is true, the difference in the cellular biochemical phenotype seems to be well represented from the clinical hematologic phenotype. In fact, while in FA patients, progressive bone marrow failure appears during the first decade of life and has an incidence of 50%−90% by 40 y of age,Citation3 in SDS, neutropenia is the most common hematologic abnormality, although anaemia and thrombocytopenia are also common. Moreover, an increased risk of developing myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML) occurs with less frequency and longer latency in SDS than in patients with FA. These differences could be due to the higher oxidative stress in FA, which also involves defects in mitochondrial structure.
The above reported observations are very interesting as they suggest how MDS represents an evolutionary outcome of very different pathological conditions. In the case of FA, the metabolic switch between glycolysis and oxidative phosphorylation might probably be associated with the process of differentiation from HSC progenitors through the modulation of calcium signals and an active communication between mitochondria and endoplasmic reticulum controlled by a modulation of the activity of the SERCA pumps.Citation41 It is interesting to note that we reported that in FA, SERCA cells are inactive,Citation33 while in SDS cells, there is a defect in calcium storage.Citation17 Calcium influx is associated with the processes of maturation and differentiation of HSC. These processes are also associated with the structural and functional maturation of mitochondria and their activation. In the case of SDS, the metabolic switches in favor of glycolysis and the progressive functional inhibition of mitochondrial activity would appear to be the result of a set of events aimed as countermeasures for the survival of cells which, for different reasons -errors in ‘RNA processing, high oxidative stress and autophagy, in the case of SDS- are in extreme danger.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Fondo Tumori e Lucemie del Bambino, AIRFA, ERG spa, Cambiaso and Risso, Rimorchiatori Riuniti, Saar Depositi Oleari Portuali, UC Sampdoria are acknowledged for supporting the activity of the Clinical and Experimental Haematology Unit of the G.Gaslini Institute. 5 per mille 2013 to IRCCS AOU San Martino – IST is acknowledged for supporting the activity of PD.
References
- Shimamura A. Inherited bone marrow failure syndromes: molecular features. Hematology Am Soc Hematol Educ Program [ Internet] 2006 [cited 2016 Jul 20]:63-71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17124042; PMID:17124042
- Dokal I, Vulliamy T. Inherited aplastic anaemias/bone marrow failure syndromes. Blood Rev 2008; 22:141-53; PMID:18164793; http://dx.doi.org/10.1016/j.blre.2007.11.003
- Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med [ Internet] 2014 [cited 2015 Dec 28]; 46:353-63. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4426964&tool=pmcentrez&rendertype=abstract; PMID:24888387
- Gonzalez-Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L. Reconsidering the Role of Mitochondria in Aging. J Gerontol A Biol Sci Med Sci [ Internet] 2015 [cited 2016 Jun 17]; 70:1334-42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25995290; PMID:25995290
- Cadenas E, Davies KJA. Mitochondrial free radical generation, oxidative stress, and aging11This article is dedicated to the memory of our dear friend, colleague, and mentor Lars Ernster (1920–1998), in gratitude for all he gave to us. Free Radic Biol Med [ Internet] 2000 [cited 2015 Mar 5]; 29:222-30. Available from: http://www.sciencedirect.com/science/article/pii/S0891584900003178; PMID:11035250; http://dx.doi.org/10.1016/S0891-5849(00)00317-8
- Dai D-F, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS. Mitochondrial oxidative stress in aging and healthspan. Longev Heal [ Internet] 2014 [cited 2015 Sep 14]; 3:6. Available from: http://www.longevityandhealthspan.com/content/3/1/6; PMID:NOT_FOUND; http://dx.doi.org/10.1186/2046-2395-3-6
- Richardson C, Yan S, Vestal CG. Oxidative stress, bone marrow failure, and genome instability in hematopoietic stem cells. Int J Mol Sci [ Internet] 2015 [cited 2016 Jun 5]; 16:2366-85. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4346841&tool=pmcentrez&rendertype=abstract; PMID:25622253; http://dx.doi.org/10.3390/ijms16022366
- Pagano G, Korkina LG, Degan P, Del Principe D, Lindau-Shepard B, Zatterale A, Franceschi C. In vitro hypersensitivity to oxygen of Fanconi anemia (FA) cells is linked to ex vivo evidence for oxidative stress in FA homozygotes and heterozygotes. Blood [ Internet] 1997 [cited 2016 Jun 17]; 89:1111-2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9028345; PMID:9028345
- Pagano G, Talamanca AA, Castello G, d'Ischia M, Pallardó F V, Petrović S, Porto B, Tiano L, Zatterale A. From clinical description, to in vitro and animal studies, and backward to patients: oxidative stress and mitochondrial dysfunction in Fanconi anemia. Free Radic Biol Med [ Internet] 2013 [cited 2016 Apr 21]; 58:118-25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23376230; http://dx.doi.org/10.1016/j.freeradbiomed.2013.01.015
- Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am [ Internet] 2013 [cited 2015 Mar 14]; 27:117-28, ix. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23351992; PMID:23351992; http://dx.doi.org/10.1016/j.hoc.2012.10.003
- Henson AL, Moore JB, Alard P, Wattenberg MM, Liu JM, Ellis SR. Mitochondrial function is impaired in yeast and human cellular models of Shwachman Diamond syndrome. Biochem Biophys Res Commun [ Internet] 2013 [cited 2015 May 25]; 437:29-34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23792098; PMID:23792098; http://dx.doi.org/10.1016/j.bbrc.2013.06.028
- Ambekar C, Das B, Yeger H, Dror Y. SBDS-deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr Blood Cancer [ Internet] 2010 [cited 2015 May 25]; 55:1138-44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20979173; PMID:20979173; http://dx.doi.org/10.1002/pbc.22700
- Pereboeva L, Westin E, Patel T, Flaniken I, Lamb L, Klingelhutz A, Goldman F. DNA damage responses and oxidative stress in dyskeratosis congenita. PLoS One [ Internet] 2013 [cited 2016 Jun 17]; 8:e76473. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24124565; PMID:24124565; http://dx.doi.org/10.1371/journal.pone.0076473
- Pereboeva L, Hubbard M, Goldman FD, Westin ER. Robust DNA damage response and elevated reactive oxygen species in TINF2-mutated dyskeratosis congenita cells. PLoS One [ Internet] 2016 [cited 2016 Jun 17]; 11:e0148793. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26859482; PMID:26859482; http://dx.doi.org/10.1371/journal.pone.0148793
- Heijnen HF, van Wijk R, Pereboom TC, Goos YJ, Seinen CW, van Oirschot BA, van Dooren R, Gastou M, Giles RH, van Solinge W, et al. Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PLoS Genet [ Internet] 2014 [cited 2016 Jan 11]; 10:e1004371. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4038485&tool=pmcentrez&rendertype=abstract; PMID:24875531; http://dx.doi.org/10.1371/journal.pgen.1004371
- Ravera S, Vaccaro D, Cuccarolo P, Columbaro M, Capanni C, Bartolucci M, Panfoli I, Morelli A, Dufour C, Cappelli E, et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie [ Internet] 2013; 95:1828-37. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23791750; PMID:23791750; http://dx.doi.org/10.1016/j.biochi.2013.06.006
- Ravera S, Dufour C, Cesaro S, Bottega R, Faleschini M, Cuccarolo P, Corsolini F, Usai C, Columbaro M, Cipolli M, et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci Rep 2016; 6:25441; PMID:27146429; http://dx.doi.org/10.1038/srep25441
- Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct [ Internet] 2012 [cited 2015 Apr 10]; 2012:646354. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3184498&tool=pmcentrez&rendertype=abstract; PMID:21977319; http://dx.doi.org/10.1155/2012/646354
- Danilova N, Sakamoto KM, Lin S. Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br J Haematol [ Internet] 2011 [cited 2016 Jun 17]; 152:217-28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21114664; PMID:21114664; http://dx.doi.org/10.1111/j.1365-2141.2010.08396.x
- Galloway CA, Yoon Y. Perspectives on: SGP symposium on mitochondrial physiology and medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol [ Internet] 2012 [cited 2015 Jun 29]; 139:455-63. Available from: http://jgp.rupress.org/content/139/6/455.full; http://dx.doi.org/10.1085/jgp.201210771
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci [ Internet] 2007 [cited 2015 Sep 14]; 120:838-48. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17298981; PMID:17298981; http://dx.doi.org/10.1242/jcs.03381
- Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta [ Internet] 2012 [cited 2015 Jun 25]; 1817:1833-8. Available from: http://www.sciencedirect.com/science/article/pii/S0005272812000692; PMID:22409868; http://dx.doi.org/10.1016/j.bbabio.2012.02.033
- Capanni C, Bruschi M, Columbaro M, Cuccarolo P, Ravera S, Dufour C, Candiano G, Petretto A, Degan P, Cappelli E. Changes in vimentin, lamin A/C and mitofilin induce aberrant cell organization in fibroblasts from Fanconi anemia complementation group A (FA-A) patients. Biochimie [ Internet] 2013 [cited 2014 Jul 15]; 95:1838-47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23831462; PMID:23831462; http://dx.doi.org/10.1016/j.biochi.2013.06.024
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan K-L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev [ Internet] 2004 [cited 2015 Jun 25]; 18:1533-8. Available from: http://genesdev.cshlp.org/content/18/13/1533.abstract?ijkey=f8958a7bbd81c783ec35dd3c909fe0893ccd3421&keytype2=tf_ipsecsha; PMID:15231735; http://dx.doi.org/10.1101/gad.1199104
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature [ Internet] 2007 [cited 2015 Jun 25]; 450:736-40. Available from: http://dx.doi.org/10.1038/nature06322; PMID:18046414; http://dx.doi.org/10.1038/nature06322
- Heiden MGV, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell [ Internet] 1999 [cited 2015 Jun 25]; 3:159-67. Available from: http://www.sciencedirect.com/science/article/pii/S109727650080307X; PMID:10078199; http://dx.doi.org/10.1016/S1097-2765(00)80307-X
- Amarachintha S, Wilson A, Pang Q. Fancd2 Deficiency Impairs Autophagy Via Deregulating The Ampk/Foxo3a/Akt Pathway. Blood 2013; 122:3713-3713; PMID:24089328; http://dx.doi.org/10.1182/blood-2013-06-508267
- Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, Berliner N, Look AT, Ebert BL, Khanna-Gupta A. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood [ Internet] 2012 [cited 2015 May 25]; 120:2214-24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3447780&tool=pmcentrez&rendertype=abstract; PMID:22734070; http://dx.doi.org/10.1182/blood-2011-10-382986
- Ibáñez A, Río P, Casado JA, Bueren JA, Fernández-Luna JL, Pipaón C. Elevated levels of IL-1beta in Fanconi anaemia group A patients due to a constitutively active phosphoinositide 3-kinase-Akt pathway are capable of promoting tumour cell proliferation. Biochem J [ Internet] 2009 [cited 2016 Jun 27]; 422:161-70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19473116; PMID:Can't; http://dx.doi.org/10.1042/BJ20082118
- Sumpter RJ, Sirasanagandla S, Fernández Á, Wei Y, Dong X, Franco L, Zou Z, Marchal C, Lee M, Clapp D, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 2016; 165:867-81; PMID:27133164; http://dx.doi.org/10.1016/j.cell.2016.04.006
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry [ Internet] 2012 [cited 2015 May 20]; 51:2959-73. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3332087&tool=pmcentrez&rendertype=abstract; PMID:22443365; http://dx.doi.org/10.1021/bi2018909
- Woods A, Dickerson K, Heath R, Hong S-P, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab [ Internet] 2005 [cited 2015 Nov 25]; 2:21-33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16054096; PMID:16054096; http://dx.doi.org/10.1016/j.cmet.2005.06.005
- Usai C, Ravera S, Cuccarolo P, Panfoli I, Dufour C, Cappelli E, Degan P. Dysregulated Ca2+ homeostasis in Fanconi anemia cells. Sci Rep [ Internet] 2015 [cited 2015 May 25]; 5:8088. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4308711&tool=pmcentrez&rendertype=abstract; PMID:25627108; http://dx.doi.org/10.1038/srep08088
- Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, Vielhaber S, Seppet E, Striggow F. The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta [ Internet] 2010 [cited 2015 May 7]; 1797:1018-27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20144582; PMID:20144582; http://dx.doi.org/10.1016/j.bbabio.2010.02.005
- Vygodina T V, Kirichenko A, Konstantinov AA. Cation binding site of cytochrome c oxidase: progress report. Biochim Biophys Acta [ Internet] 2014 [cited 2015 Sep 28]; 1837:1188-95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24607866; PMID:24607866; http://dx.doi.org/10.1016/j.bbabio.2014.02.025
- Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett [ Internet] 2004 [cited 2015 May 16]; 567:96-102. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15165900; PMID:15165900; http://dx.doi.org/10.1016/j.febslet.2004.03.071
- Chotinantakul K, Leeanansaksiri W. Hematopoietic stem cell development, niches, and signaling pathways. Bone Marrow Res [ Internet] 2012 [cited 2016 Jul 20]; 2012:270425. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22900188; PMID:22900188; http://dx.doi.org/10.1155/2012/270425
- Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science [ Internet] 2015 [cited 2015 Oct 7]; 348:340-3. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4405120&tool=pmcentrez&rendertype=abstract; PMID:25837514; http://dx.doi.org/10.1126/science.1260384
- Kitsos CM, Sankar U, Illario M, Colomer-Font JM, Duncan AW, Ribar TJ, Reya T, Means AR. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J Biol Chem [ Internet] 2005 [cited 2016 Jun 17]; 280:33101-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16020540; PMID:16020540; http://dx.doi.org/10.1074/jbc.M505208200
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature [ Internet] 2015 [cited 2015 Feb 18]; 520:549-52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25707806; PMID:25707806; http://dx.doi.org/10.1038/nature14131
- Papp B, Brouland J-P, Gélébart P, Kovàcs T, Chomienne C. Endoplasmic reticulum calcium transport ATPase expression during differentiation of colon cancer and leukaemia cells. Biochem Biophys Res Commun [ Internet] 2004 [cited 2016 Jun 22]; 322:1223-36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15336970; PMID:15336970; http://dx.doi.org/10.1016/j.bbrc.2004.08.030