
ABSTRACT
We have examined the relationship between checkpoint adaptation (mitosis with damaged DNA) and micronuclei. Micronuclei in cancer cells are linked to genomic change, and may induce chromothripsis (chromosome shattering). We measured the cytotoxicity of the cancer drug cisplatin in M059K (glioma fibroblasts, IC50 15 μM). Nearly 100% of M059K cells were positive for histone γH2AX staining after 48 h treatment with a cytotoxic concentration of cisplatin. The proportion of micronucleated cells, as confirmed by microscopy using DAPI and lamin A/C staining, increased from 24% to 48%, and the total micronuclei in surviving cells accumulated over time. Promoting entry into mitosis with a checkpoint inhibitor increased the number of micronuclei in cells whereas blocking checkpoint adaptation with a Cdk inhibitor reduced the number of micronuclei. Interestingly, some micronuclei underwent asynchronous DNA replication, relative to the main nuclei, as measured by deoxy-bromo-uracil (BrdU) staining. These micronuclei stained positive for histone γH2AX, which was linked to DNA replication, suggesting that micronuclei arise from checkpoint adaptation and that micronuclei may continue to damage DNA. By contrast the normal cell line WI-38 did not undergo checkpoint adaptation when treated with cisplatin and did not show changes in micronuclei number. These data reveal that the production of micronuclei by checkpoint adaptation is part of a process that contributes to genomic change.
Introduction
Tumors that are resistant to treatment frequently have a complex genome organization. The Cancer Genome Sequencing Atlas project has sequenced tumor genomes from hundreds of patients with solid tumors such as colorectalCitation1 or brain cancers.Citation2 The tumor from each patient presented a different DNA sequence and chromosome organization, highlighting that genomic complexity likely contributes to the difficulty in providing effective treatments. Because most tumors differ from each other, it is likely that an effective cancer treatment will require more than one type of chemotherapeutic approach. A better understanding of how genomic complexity arises is needed to provide insights to treating cancer patients successfully.
One way cells can acquire genomic change is through checkpoint adaptation.Citation3 Checkpoint adaptation is characterized by a cell-cycle arrest after detection of damaged DNA, overcoming this arrest, and entry into mitosis with damaged DNA.Citation4,5 The presence of damaged DNA is signaled by cells by activating histone γH2AX,Citation6 which leads to a series of pathways steps including the activation of DNA damage kinases ATM/ATR, which then phosphorylate checkpoint kinase 1 (Chk1) on serine 345.Citation7 Chk1 is a protein kinase that prevents the activation of the mitotic enzyme Cdk1 by increasing the activity of Wee1Citation8 and inhibiting Cdc25C.Citation9 By this pathway, cells arrest in G2-phase with 4N DNA. Although it was believed that cells would die by apoptosis when challenged by damaged DNA, cells have the possibility to engage another pathway known as checkpoint adaptation.Citation10 During checkpoint adaptation, cells enter mitosis despite having damaged DNA.Citation11,12 In these cells, the activity of Chk1 decreases, thus permitting the activation of Cdk1 followed by entry into mitosis damaged DNA.
The outcomes of checkpoint adaptation are not yet precisely understood. Although most cells that undergo checkpoint adaptation die, a small yet biologically significant number of cells survive.Citation10,12 Because these cells have undergone mitosis with damaged DNA, it is likely they acquire changes to their genome.Citation13 One genomic change that characterizes cancer cells is the presence of micronuclei. Micronuclei form from lagging whole chromosomesCitation14,15 or chromosome fragmentsCitation16 that are not incorporated into the main nucleus following anaphase.Citation17 This can occur if the centromere is either damagedCitation18 or missingCitation19 as is the case for acentric chromosome fragments. In addition, damaged chromosomes may fuse and create a dicentric chromosomeCitation17 that becomes attached to both spindle poles. Dicentric chromosomes form a nucleoplasmic bridge (NPB) between daughter nuclei after anaphase. The NPB is subsequently broken in cytokinesis and the resulting DNA fragments are then incorporated into micronuclei. Many studies have demonstrated that genotoxic cancer treatments can induce micronuclei.Citation20
The relationship between micronuclei and checkpoint adaptation is not well understood. Here we examine whether the formation of micronuclei requires checkpoint adaptation. We focused our study on M059K cells, a cancer cell line that has previously been reported to undergo checkpoint adaptation in response to pharmacological concentrations of CPT.Citation12 Furthermore, these cells have also been reported to form micronuclei after a genotoxic treatment.Citation21 We also examine normal lung fibroblasts to test if checkpoint adaptation or micronuclei formation can occur in normal cells. The results of this study will help us understand how treated cancers cells that survive checkpoint adaptation acquire complex genomic changes.
Results
Characterization of micronuclei in glial cancer cells
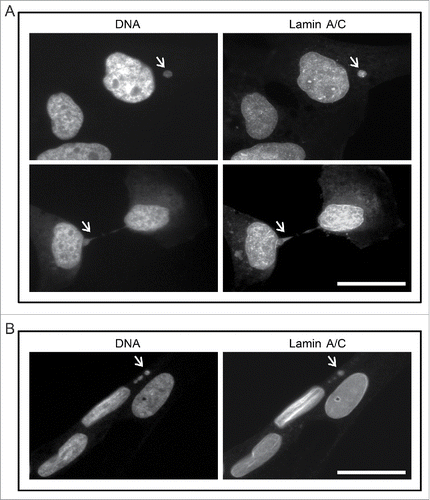
We stained M059K cells with DAPI and with anti-lamin A/C antibodies and observed them by immunofluorescence microscopy to count micronuclei (). In an exponentially growing culture, 25% ± 1% of cells contained at least one extra-nuclear structure in addition to the main nucleus. A much lower number of cells had nucleoplasmic bridges (0.5% ± 0.1%). We confirmed that 94% ± 8% of micronuclei identified by DAPI also were positive for lamin A/C staining. Using the same protocol, we examined normal human lung fibroblasts (WI-38) for micronuclei. By contrast to M059K cells, only 2% ± 2% of WI-38 cells contained DAPI positive extra nuclear structures of which 47% ± 5% were lamin A/C positive. No nucleoplasmic bridges were detected. These results confirmed that M059K cells have micronuclei, and that micronuclei are predominantly found in cancer cells.
Figure 1. Human glioma cells contain micronuclei and nucleoplasmic bridges. (A) M059K cells were stained for either DNA (left) or lamin A/C (right) and observed by immunofluorescence microscopy. Arrows indicate a micronucleus (top) or a nucleoplasmic bridge (bottom). Scale bar = 25 µm. (B) Non-cancerous human lung fibroblasts (WI-38) were stained for either DNA (left) or lamin A/C (right) and observed by immunofluorescence microscopy. The nuclei of 3 cells are shown. Arrows indicate a micronucleus. Scale bar = 25 µm.
Characterization of genotoxic agents to induce micronuclei
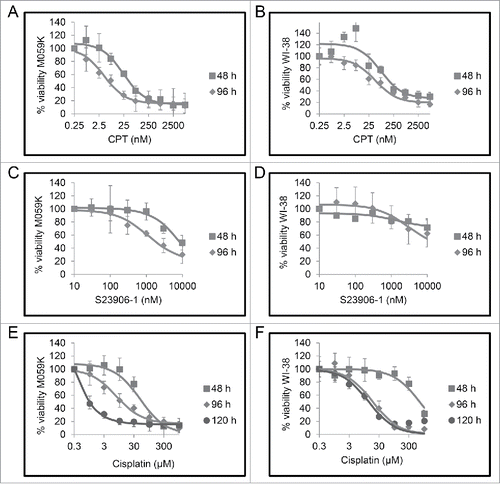
To test the relationship between checkpoint adaptation and micronuclei in cancer cells, we first selected a genotoxic agent for use in our experiments. Our criteria for selection were: an agent that has an IC50 concentration to cultured cells similar to pharmacological concentrations, and a difference in cell viability between early and late times after treatment, which is a feature of checkpoint adaptation.Citation3 We tested camptothecin (CPT), S23906-1, and cisplatin because each has a different mechanism of action. Increasing concentrations of either CPT, S23906-1, or cisplatin were added to cells and viability was measured at 48, 96, or 120 hours by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (). We observed that cell viability was reduced in both M059K and WI-38 cells as the concentration of each genotoxic agent was increased. The greatest difference in cell viability between 48 and either 96 or 120 hours in both cell lines occurred in the cisplatin treated group (). Furthermore, the cisplatin treated group had IC50 values of 1 μM at 120 hours to M059K cells and 18.5 μM to WI-38 cells, values that are clinically relevant.Citation3 Although S-23906 and CPT were both cytotoxic to M059K cells, WI-38 cells were either insensitive to the compounds as in the case of S23906-1, or only sensitive to concentrations above the pharmacological concentrations (). Based upon these tests, we selected cisplatin as the genotoxic agent to investigate the relationship between checkpoint adaptation and micronuclei.
Figure 2. Cells treated with increasing concentrations of genotoxic agents have reduced cell viability. M059K and WI-38 cells were treated with increasing concentrations of either camptothecin (CPT) A and B, S23906-1 C and D, or cisplatin E and F for either 48 h (squares), 96 h (diamonds), or 120 h (circles). Cell viability was measured using the MTT assay. Means of 3 experiments and standard errors of means are shown.
Cisplatin induces micronucleation in M059K cells
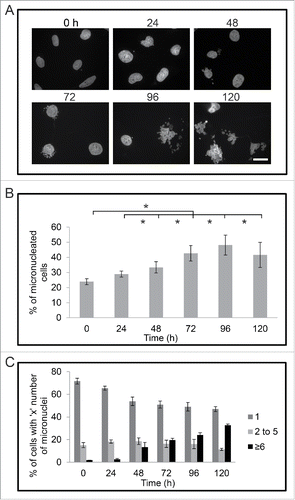
We tested if treatment with cisplatin would increase either the number of cells that had micronuclei, or the number of micronuclei per cell. M059K cells were treated with 30 μM cisplatin for times up to 120 h and then stained with DAPI and observed by immunofluorescence microscopy (). The number of cells with micronuclei increased over time after treatment with cisplatin (). At time 0, the percentage of cells with micronuclei was 24% ± 2%, but this percentage doubled to 48% ± 7% by 96 hours. We also observed an increase in the number of micronuclei per cell after treatment (). At time 0, 72% ± 2% of the micronucleated cells had one micronucleus while only 2% ± 0% of M059K cells had 6 or more micronuclei. The percentage of cells containing 6 or more micronuclei increased to 13% ± 4% by 48 h and continued to increase to 33% ± 1% by 120 h of treatment. The number of cells containing 2 to 5 micronuclei per cell did not change in a significant manner during the treatment.
Figure 3. Cisplatin treatment increases the proportion of micronucleated M059K cells. (A) Cells were fixed at indicated times (hours), stained for DNA, and observed by immunofluorescence microscopy. Scale bar = 25 μm. (B) The mean percentage of micronucleated cells for each time point of cisplatin treatment is shown. Standard error of means are shown. Asterisks show significant difference, p < 0.05. (C) Mean percentage of micronuclei per cell is shown. Standard error of means are shown. Asterisks show significant difference, p < 0.05.
Micronucleated cells persist after treatment with cisplatin
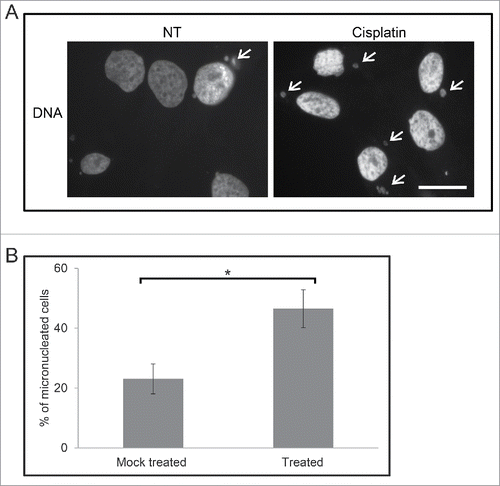
We treated M059K cells with 30 μM cisplatin for 120 h, and then cultured them for additional 8 to 10 d without cisplatin. Although the majority of cells had died, as expected, cells that survived were stained with DAPI and observed by immunofluorescence microscopy (). The percentage of survival cells with micronuclei was 47% ± 6% compared to 23% ± 5% in the mock treated population (p < 0.05). Interestingly, the percentage of survival micronucleated cells was similar to those of cells analyzed immediately after 120 h treatment. Together, these data confirm that exposure to cisplatin causes a large increase in the number of micronuclei and these micronuclei are maintained in cells that survive the treatment.
Figure 4. M059K cells retain micronuclei after treatment with cisplatin. (A) Cells were either non-treated (NT) or treated with 30 µM cisplatin for 120 h and then cultured for 8 to 10 d. Cells were stained to mark DNA and then observed by immunofluorescence microscopy. Arrows indicate micronuclei. Scale bar = 25 μm. (B) The mean percentage of M059K cells either mock treated (all steps but no cisplatin) or treated with 30 µM cisplatin for 120 h and then cultured for 8 to 10 d was calculated. Standard error of means are shown. Asterisk shows significant difference, p < 0.05.
Micronuclei arise in M059K cells that have undergone checkpoint adaptation
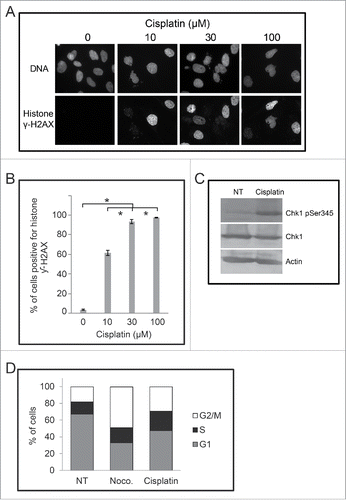
To determine if the increase in the number of micronuclei in cisplatin treated cells was linked to checkpoint adaptation, we first examined cells for damaged DNA following treatment. Cells were treated with increasing concentrations of cisplatin (0 to 100 μM) and then stained with DAPI and anti-histone γH2AX antibodies to detect DNA and damaged DNA, respectively (). The number of cells positive for histone γH2AX increased from 2% in non-treated cells to 61% ± 3% by 10 μM cisplatin, 94% ± 2% by 30 μM cisplatin and 97% with 100 μM cisplatin (p < 0.05) ). We found that the cells treated with 30 μM cisplatin for 24 h contained relatively higher levels of phospho-ser 345 checkpoint kinase 1 (Chk1) whereas the total amount of Chk1 did not change (). We then examined cisplatin treated cells by flow cytometry to detect DNA content. Cells were either non-treated, treated with 200 ng/mL of nocodazole (M-phase arresting agent), or treated with 30 μM cisplatin and analyzed at 24 h (). Non-treated cells were predominantly in the G1 phase (67% ± 1%) with the remaining cells in either S phase (14.6 ± 1%) or G2/M phase (18% ± 2%). By contrast, the population treated with 30 µM cisplatin had 24% cells in S phase and 30% cells in G2/M phase; nocodazole treated culture had 49% in the G2/M-phase (p < 0.05). These data revealed that cisplatin treatment damages DNA, induces the phosphorylation of Chk1, and causes the cells to arrest in the cell cycle, which are prerequisites for checkpoint adaptation.Citation4
Figure 5. Cells signal damaged DNA in a dose-dependent manner following treatment with cisplatin. (A) M059K cells were treated with increasing concentrations of cisplatin for 48 hours and then stained for either DNA (top row) or histone γH2AX (bottom row) and observed by immunofluorescence microscopy. Scale bar = 50 µm. (B) The mean proportion of cells positive for histone γH2AX was determined for each treated population. Asterisks show significant differences, p < 0.05. (C) Protein extracts were prepared from M059K cells that were either non-treated or treated with 30 µM cisplatin for 24 hours. Samples were processed by western blotting with antibodies against either phospho-S345 Chk1, Chk1, or actin. (D) M059K cells were either non-treated, treated with 200 ng/mL nocodazole, or 30 µM cisplatin for 24 h and then analyzed by flow cytometry. DNA content was determined with propidium iodine staining and the mean percentage of cells in a cell cycle phase was estimated from 2 experiments.
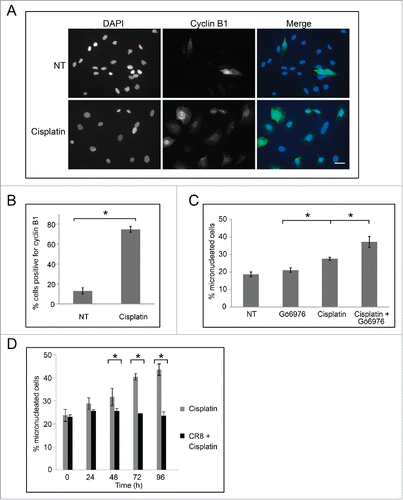
We next examined cells for cyclin B1 staining by immunofluorescence microscopy (). In non-treated cell populations, only 13% ± 3% of cells were cyclin B1 positive. By contrast, treatment with 30 μM cisplatin increased the percentage of cyclin B1 positive cells to 75% ± 3% (p < 0.05) (), revealing that the treated cells were prepared to undergo checkpoint adaptation. We next tested whether addition of an inhibitor of Chk1, Gö6976, would affect the number of micronuclei in cells. Cells were either non-treated, treated with 15 nM Gö6976 for 24 h, 30 μM cisplatin for 48 h, or with 30 μM cisplatin for 24 h and then co-treated with 15 nM Gö6976 for an additional 24 h. Cells were stained with DAPI and then observed by immunofluorescence microscopy (). No change in the number of micronuclei was observed between the non-treated and Gö6976 treated cells (p = 0.05), and treatment with cisplatin alone led to a significant increase in micronuclei. By contrast, co-treatment of cisplatin and Gö6976, resulted in an increase in cells with micronuclei from 28% ± 1% to 38% ± 3% (p < 0.05) (). These data demonstrate that bypassing the DNA damage checkpoint through the inactivation of Chk1 results in a larger number of micronucleated cells. We then performed the complementary experiment by inhibiting mitosis and asking what would be the outcome upon micronucleated cell number. Cells were treated with either 30 μM cisplatin or co-treated with 30 μM cisplatin and the Cdk inhibitor, CR8 (). As had been shown, treatment with cisplatin increased the percentage of micronucleated cells; by contrast, no change in the number of micronucleated cells was observed in populations co-treated with 30 μM cisplatin and CR8.
Figure 6. Cells treated with cisplatin undergo checkpoint adaptation and acquire additional micronuclei. (A) M059K cells were either non-treated (top row) or treated with 30 μM cisplatin for 48 h, and all cells were stained with DAPI and anti-cyclin B1 antibodies, then observed by immunofluorescence microscopy. Merge of images is shown on right. Scale bar equals 50 μm. (B) The mean percentage of cells positive for cyclin B1 was calculated. Standard error of the mean is shown. Asterisk shows significant difference. p < 0.05. (C) M059K cells were either non treated, treated with 15 nM Gö6976 for 24 h, 30 μM cisplatin for 48 h, or 30 μM cisplatin for 24 h and then co-treated with 15 nM Gö6976 for an additional 24 h. The mean percentage of micronucleated cells was calculated for each treatment and standard error of means are shown. Asterisks show significant differences. p < 0.05. (D) M059K cells were either non-treated, treated with 30 μM cisplatin, or co-treated with cisplatin and 500 nM CR8 for indicated times. The mean percentage of micronucleated cells and standard error of means are shown. Asterisks show significant differences. p < 0.05.
Micronuclei undergo DNA replication that damages DNA
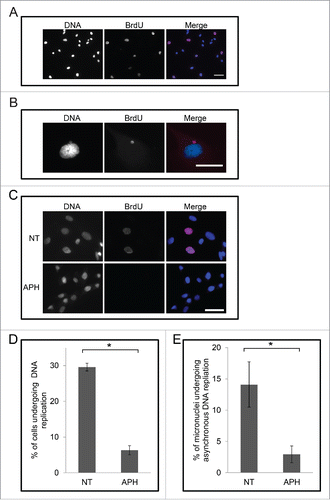
Having demonstrated that cells that survive treatment by cisplatin acquire micronuclei we then examined these micronuclei for their capacity to undergo DNA replication. M059K cells were examined for the incorporation of the nucleoside analog 5-bromo-2′-deoxy-uridine (BrdU) under various conditions relevant to checkpoint adaptation. Cells were incubated with 30 μM BrdU and then stained with DAPI and an anti-BrdU antibody, and observed by immunofluorescence microscopy (). We detected BrdU incorporation in the main nuclei in 35% ± 4% of non-treated cells (). Strikingly, however, we observed that BrdU signals in micronuclei and main nuclei were not correlated in 12% ± 7% of non-treated cells (). For example, BrdU signals were present either in the micronucleus and not in the main nucleus, or present in the main nucleus and not the micronucleus. To confirm the BrdU experiments, we also labeled cells with 5-ethynyl-2′-deoxyuridine (EdU) and obtained similar results (not shown). We verified that the BrdU signals were due to DNA replication by applying the DNA polymerase I α inhibitor, aphidicolin. We treated M059K cells with 10 μg/mL aphidicolin for 24 h, added BrdU to the media, and then examined the incorporation of BrdU by immunofluorescence microscopy (, ). In the presence of aphidicolin only 5% ± 1% of cells had incorporated BrdU (p < 0.05). Knowing that aphidicolin could block DNA synthesis in this cell model, we then examined BrdU incorporation in micronuclei in the presence of aphidicolin. Aphidicolin reduced BrdU incorporation of micronuclei in cells from 14% ± 3% to 3% ± 1%, and EdU incorporation from 18% to 5%. These data reveal that micronuclei undergo DNA replication, and it can occur at times different from those of the main nucleus.
Figure 7. Micronuclei in M059K cells undergo asynchronous DNA replication relative to the main nucleus. (A) Cells were incubated with 30 μM BrdU for 30 min then fixed and stained for either DNA (left) and BrdU (middle) and then observed by immunofluorescence microscopy. Merge of images is shown on right. Scale bar = 50 μm. (B) Cells were processed as described in A. An image of a main nucleus and a micronucleus is shown. Scale bar = 25 μm. (C) Cells were treated either non-treated or treated with 10 μg/mL aphidicolin for 24 h and 30 μM BrdU for the final 30 min, then stained for either DNA (left) or BrdU (middle), and then observed by immunofluorescence microscopy. Merge of images is shown on the right. Scale bar = 50 μm. (D) Cells from images collected in C were counted and the mean percentage of cells undergoing positive for BrdU staining is shown. Asterisk shows significant difference. p < 0.05. (E) Cells from images collected in (C) were counted and the mean percentage of micronuclei that were positive for BrdU staining in the absence of main nucleus staining is shown. Asterisk shows significant difference. p < 0.05.
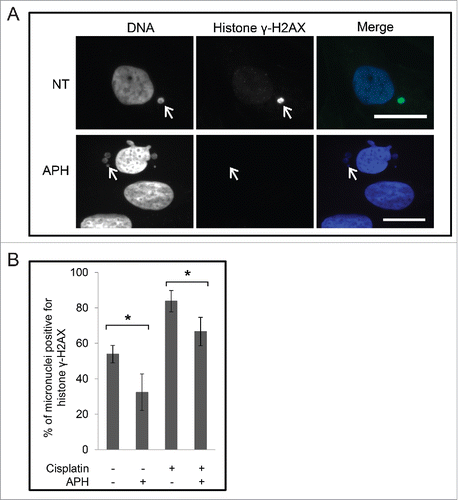
Unscheduled DNA replication is a source of genomic instability in cells; therefore we examined micronuclei in M059K cells for damaged DNA by histone γH2AX staining. In non-treated cells 54% ± 5% of micronuclei were positive for histone γH2AX staining despite that the main nucleus was negative for histone γH2AX staining (). When cells were treated with 10 μg/mL aphidicolin and then stained with a histone γH2AX antibody we observed a reduction to 32% ± 10% (p < 0.05) in the number of micronuclei that were positive for histone γH2AX. We then tested whether additional micronuclei would be damaged by treatment with cisplatin. Cells were either non-treated, treated with 30 μM cisplatin for 48 h, or with 30 μM cisplatin for 24 h and then co-treated with aphidicolin for an additional 24 h. Cells were then stained with DAPI and an anti-histone γH2AX antibody and observed by immunofluorescence microscopy (). Under these conditions, 83% ± 6% of micronuclei contained histone γH2AX signals after treatment with cisplatin. This value was reduced to 67% ± 8% in cells co-treated with cisplatin and aphidicolin (p < 0.05). These data confirm that micronuclei harbor damaged DNA that likely originated from DNA replication, and that micronuclei can acquire additional damage by treatment with cisplatin.
Figure 8. Micronuclei are positive for histone γH2AX in the absence of a genotoxic agent. (A) Cells were either non-treated (NT) or treated with aphidicolin for 24 h, and were stained with DAPI and anti-histone γH2AX antibodies, and observed by immunofluorescence microscopy. Merge of images is shown on right. Scale bar = 25 μm. The arrows point to micronuclei. (B) Images cells treated as described in (A), and treated with cisplatin, or with cisplatin and aphidicolin were counted and the mean percentages of micronuclei positive for histone γH2AX staining are shown. Standard error of means are shown. The asterisks show significant differences.
Normal cells treated with cisplatin do not undergo checkpoint adaptation or form micronuclei
Having identified a relationship between checkpoint adaptation and micronuclei formation in M059K cells, we investigated if the normal, WI-38 cells could undergo checkpoint adaptation. WI-38 cells were either non-treated or treated with increasing concentrations of cisplatin (10 μM to 300 μM) for 48 h, stained with DAPI and an anti-histone γH2AX antibody, then observed by immunofluorescence microscopy (). The percentage of WI-38 cells positive for histone γH2AX staining at the cytotoxic concentration of 30 μM cisplatin was 79% ± 3%, and it increased to 95% with treatments of 100 μM or greater (p < 0.05). At concentrations lower than or equal to 30 μM, cells had histone γH2AX foci but at concentrations greater than 30 μM cisplatin, pan nuclear staining was observed. Non-treated WI-38 cells did not stain for histone γH2AX. We prepared protein extracts from WI-38 cells that were either non-treated or treated with 30 μM cisplatin for 24 h and examined them by western blotting for Chk1 and phospho-Ser345 Chk1 (). Phospho-Ser345-Chk1 was absent in non-treated cells and present in cisplatin treated cells, which coincided with an increase in total Chk1 levels as equal protein amounts were loaded. Although WI-38 cells signaled damaged DNA and contained phospho-Ser345-Chk1, they did not display a specific cell cycle arrest, as determined by flow cytometry (). We then compared the number of WI-38 cells that expressed cyclin B1 protein in non-treated and in cells treated with 30 μM cisplatin (). Only 13% ± 1% of non-treated cells and 22% ± 1% of cells treated with 30 μM cisplatin were positive for cyclin B1 (p < 0.05).
Figure 9. Normal cells do not undergo checkpoint adaptation after cisplatin treatment and do not acquire micronuclei. (A) Cells were treated with increasing concentrations of cisplatin for 48 h, stained for histone γH2AX and observed by immunofluorescence microscopy. The number of cells with nuclear histone γH2AX staining was counted for each treatment and the mean percentage of positive was calculated with standard error of means. Asterisks show significant differences. p < 0.05. (B) Protein extracts were prepared from WI-38 cells that were either non-treated or treated with 30 µM cisplatin for 24 hours. Samples were processed by western blotting with antibodies against either phospho-S345 Chk1, Chk1, or actin. (C) WI-38 cells were either non-treated, treated with 200 ng/mL nocodazole, or 30 µM cisplatin for 24 h and then analyzed by flow cytometry. DNA content was determined with propidium iodine staining and the mean percentage of cells in a cell cycle phase was estimated from 2 experiments. (D) WI-38 cells were either non-treated or treated with 30 μM cisplatin for 48 hours, and all cells were stained with DAPI and anti-cyclin B1 antibodies, then observed by immunofluorescence microscopy. The mean percentage of cells that were positive for cyclin B1 either group was calculated. E. WI-38 cells were treated with 30 μM cisplatin for up to 120 h and then fixed at indicated times, stained for DNA, and observed by immunofluorescence microscopy. The mean proportion of micronucleated cells was determined and compared over time. Standard error of means are shown.
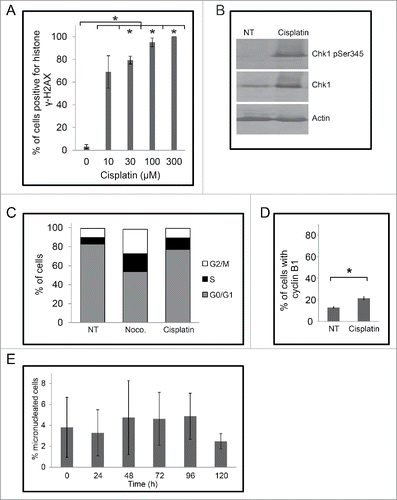
We then examined WI-38 cells for micronuclei after treatment by 30 μM cisplatin for times up to 120 h. Cells were stained with DAPI, and observed by immunofluorescence microscopy and micronuclei were counted. By 72 h, we observed dying cells with discrete, brightly-stained chromatin structures. During the timecourse, the number of micronuclei in cells did not change ().
Discussion
The Cancer Genome Atlas (TCGA) sequenced the genomes from glioblastoma tumors isolated from 206 patients and reported that each tumor had a different sequence from that of any other tumor.Citation2 The sequence differences included nucleotide substitutions, amplifications, deletions, and chromosomal rearrangements. In a follow-up study, aneuploidy was identified in the genomes of tumors isolated from another cohort of 128 glioblastoma patients.Citation22 The origin of this profound genomic heterogeneity is not yet known, but the heterogeneity likely contributes to the difficulty in providing effective treatment options for glioblastoma patients.
M059K cells have features that make them a convenient model in which to study the relationship between micronuclei and checkpoint adaptation. These cells are able to undergo checkpoint adaptation when exposed to a variety of genotoxic agents.Citation12 Furthermore, like other glioma and neuroblastoma cell lines, they naturally have a relatively high number of micronuclei, which provides sufficient material for counting assays.Citation23 We confirmed the presence of micronuclei in M059K cells by using lamin A/C staining to mark extra-nuclear, DNA containing structures. Typically, M059K cells have 65 to 79 chromosomes and at least 22% of cells exhibit polyploidy. In addition, these cells have several complex chromosomal rearrangements, including reciprocal and non-reciprocal translocations.Citation24 Although the precise mechanism is not known, it is possible that the absence of functional p53 enables cells to tolerate genomic change better than cells with p53.Citation25 Variability in chromosome number and genomic rearrangements are frequently associated with the presence of micronuclei.Citation17
We selected cisplatin as a genotoxic agent to treat M059K cells because we were able to achieve cytotoxicity at concentrations in culture conditions that reflected serum concentrations in treated patients.Citation3 Concentrations that exceed 40 µM are reported to cause liver and kidney failure, hearing loss, pancytopenia (reduction of blood cells), and death in human patients Citation26 and are therefore not pharmacologically relevant.Citation27 Importantly, 80% of M059K cells and 90% of WI-38 cells that were alive at 48 h had died by 120 h treatment with 30 µM cisplatin. The delay in the loss of cell viability between 48 h and 120 h is typical of cells that undergo cell death at pharmacological concentrations of anti-cancer drugs.Citation12 By contrast if the concentration of cisplatin applied to cells was at supra-pharmacological levels (100-300 µM), cells died within 48 h and do not undergo checkpoint adaptation.Citation10
M059K cells treated with cisplatin undergo checkpoint adaptation and form micronuclei
We observed 3 changes in M059K cells that survived cisplatin treatment: 1) an increase in the number of cells with micronuclei; 2) an increase in the number of micronuclei per cell; and 3) an increase in nuclearplasmic bridges. These observations are consistent with those of studies that reported that cisplatin induces micronuclei formation in other cancer epithelial cells such as HeLa and HT1080.Citation28,29 We also observed that cells retained micronuclei 8 to 10 d post treatment.
One pathway that allows entry into mitosis with damaged DNA is checkpoint adaptation. This pathway is characterized by 3 steps: 1) DNA damage induces cell cycle arrest; 2) overcoming this arrest; 3) and entry into mitosis with damaged DNA.Citation4 Chk1 and Cdk1 are required for this process, which upon activation, control the arrest in G2-phase and entry into mitosis, respectively.Citation3 M059K cells treated with cisplatin demonstrated the key steps of checkpoint adaptationCitation12,30: they signaled damaged DNA by histone γH2AX, activated Chk1, arrested in the cell cycle, and the majority of cells expressed cyclin B1. With this information we could then test the prediction that the inhibition of Chk1 would cause more cells to enter mitosis with damaged DNA and therefore generate additional micronuclei.Citation31 We observed that the percentage of micronuclei in cells co-treated with a checkpoint inhibitor and cisplatin was higher than the percentage of cells with micronuclei after treatment with cisplatin alone. This finding suggested that inhibiting Chk1 permits entry into mitosis,Citation32 which generates additional micronuclei. Our observations are consistent with those of Petsalaki et al. who reported that Chk1-deficient human colon carcinoma cells (BE) form additional micronuclei, as do cells treated with etoposide.Citation20,33 We then found that cells co-treated with the Cdk1 inhibitor CR8 and cisplatin had a similar number of micronuclei as cells that were not treated with cisplatin. Our findings provide evidence that checkpoint adaptation is linked to the formation of micronuclei.
Micronuclei cause additional damage to DNA
It had been previously reported that micronuclei with lamina, such as those identified by this study, are competent to undergo DNA replication.Citation34 We were able to identify DNA replication in micronuclei in experiments in which we measured BrdU and EdU incorporation. Strikingly, we observed micronuclei that underwent DNA replication asynchronously relative to that of the main nucleus. We confirmed that BrdU and EdU signals were dependent upon DNA polymerase activity by co-treating cells with aphidicolin, a DNA polymerase inhibitor.Citation35,36 The precise reason for asynchronous DNA replication is not known, although it has been observed in several cell types.Citation37,38 It is proposed that micronuclei may have poorly functioning membrane,Citation39 which would affect the replication by a variety of means including DNA replication protein import and DNA organization.Citation40 In addition, if the DNA is damaged in the micronucleus, then DNA replication might be delayed, and if it is delayed to the point when the cell undergoes mitosis, DNA could be further damaged.
Nearly half of the micronuclei in M059K cells were positive for histone γH2AX, even in the absence of cisplatin treatment. We confirmed that the histone γH2AX staining was dependent upon DNA replication as cells treated with aphidicolin (in the absence of cisplatin) had fewer micronuclei positive for histone γH2AX. Our observations extend those of Crasta et al. who reported that 60% of micronuclei produced by lagging chromosomes were positive for histone γH2AX staining.Citation15 Our results also suggest that micronuclei are sources of continuous DNA damage, which may be caused by undergoing DNA replication at different times from that of the main nucleus. Micronuclei can undergo massive genomic change by the process of chromothripsis,Citation40 in which checkpoint adaptation might be a key step to start this process. Furthermore, chromothripsis is more likely to be identified in human cells that are mutated for p53.Citation25 It is possible that the absence of p53 maybe required to tolerate or even survive such types of genomic change. In the yeast, disruption of S-phase and checkpoint bypass can lead to micronuclei formation and genomic change.Citation38
Checkpoint adaptation and micronuclei are rare in normal WI-38 cells
We could not detect signs of checkpoint adaptation in WI-38 cells treated by conditions similar to those used on M059K cells. We noted that WI-38 cells signaled damaged DNA by histone γH2AX and had activated Chk1. The absence of change micronuclei number in this cell line was consistent with the lack of observed checkpoint adaptation in these cells. The engagement of checkpoint adaptation in normal, non-cancerous cell lines appears to be more variable than that of tumor cells lines. Checkpoint adaptation has been observed in normal MEF or BJ cells exposed to radiation.Citation13,41 Yeast are also capable of undergoing checkpoint adaptation.Citation4 In experiments using other genotoxic agents, normal fibroblasts did not acquire micronuclei after exposure to neocarzinostatin,Citation42 or hydroxyurea.Citation43 By contrast to M059K cells, which have a mutated, non-functional version of p53,Citation44 WI-38 cells have a functional protein p53, and depletion of p53 in WI-38 cells leads to micronuclei in cells.Citation42 The type of damaged DNA may affect if normal cells engage checkpoint adaptation or not. By cisplatin treatment, WI-38 cells arrest largely in G1 and few cells expressed cyclin B1 protein, which would be critical for activation of Cdk1 and entry into mitosis. There are numerous genetic and experimental examples of how inhibition of Chk1 protein leads to entry into mitosisCitation45,46 or micronuclei formation.Citation42 It is noteworthy, however, that the simplest approach such as re-introducing functional p53 in a cancer cell line does not prevent mitotic catastrophe, or likely checkpoint adaptation.Citation47 In the absence of the intact DNA damage repair and checkpoint system, cancer cells undergo checkpoint adaptation, or in the presence of Chk1 inhibitors, glioma cells can enter mitosis with damaged DNA.Citation48 We find that checkpoint adaptation lead to the production of micronuclei, which in turn, contributes to a continuous process of genomic change in cancer cells.
Materials and methods
Cell culture
Human M059K and WI-38 cells were obtained from the American Type Culture Collection (CRL-2365; CCL-75). M059K cells were maintained in Dulbecco's Modified Eagle medium (DMEM) F-12 (Gibco; 11320-082), supplemented with 10% heat inactivated fetal calf serum (Gibco; 12484028), 2 mM non-essential amino acids (Gibco; 11140050), and 15 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4. WI-38 cells were maintained in DMEM/high glucose (Sigma; D6546) supplemented with 10% heat inactivated fetal calf serum (Gibco; 12484028), 2 mM non-essential amino acids (Gibco; 11140050), and 1.6 mM GlutaMAX (Gibco; 35050-061). Cells were grown at 37°C in 5% CO2 and media was changed every 3 to 4 d. M059K and WI-38 cells were plated at a density of 5.0 × 105 cells/75 cm2 flask and cultured for 48 h prior to treatment. Cisplatin (Sigma; 479306-1G) was dissolved in dimethyl sulfoxide (DMSO) (Sigma; D2438) to a concentration of 100 mM. CR8 (Tocris Biosciences; 2706/10), Gö6976 (Tocris Biosciences; 2253), CPT (Sigma; 7689-03-4) and S23906-1 (Servier) were dissolved to a concentration of 10 mM, respectively. Nocodazole (Sigma-Aldrich; M1404-10MG), and aphidicolin (Santa Cruz; sc-201535) were dissolved in DMSO at concentrations of 200 μg/mL and 10 mg/mL respectively. All compounds were stored at −20°C until use.
Cytotoxicity assays
Cytotoxicity of CPT, S23906-1, and cisplatin was measured by the microculture tetrazolium assay (MTT; (3-(4,5)-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide).Citation49 M059K or WI-38 cells were cultivated at 4,000 cells per well on a 96 well plate for 48 hours prior to treatment. Results were expressed as IC50, which was the compound concentration that reduced the absorbance by 50% at 590 nm, compared to DMSO treated cells. All measurements were done in triplicate.
Light microscopy
M059K cells were cultivated at 50,000 cells per well on a 6 well plate for 48 hours prior to treatment. Images were collected at 24 hours after treatment with an Infinity 1.5 camera powered by Infinity Capture (Lumenera Corporation) software on an Olympus BX41 inverted microscope.
Flow cytometry
Total cultures were collected by trypsinization. Cells were washed in phosphate-buffered saline (PBS) and fixed in 90% ethanol (−20°C) for at least 24 h. For analysis, samples were incubated for 20 minutes in wash buffer with 0.02 mg/mL propidium iodide (Invitrogen) and 0.2 mg/mL RNAse A (Sigma), and analyzed by a FACS Canto II flow cytometer (BD Biosciences) using BD FACSDiva software. Gating was set using control samples of cells treated with either 25 nM CPT or 200 ng/mL nocodazole.Citation12 Experiments were repeated twice.
Extract preparation
Cells were trypsinized, passed through a 26 gauge needle 5 times, and resuspended in extraction buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 50 mM NaF, 10 mM EGTA (ethyleneglycol-bis(β-aminoethylether)-N,N′-tetraacetic acid), 50 mM β-glycerophosphate, 1 mM ATP, 1 mM (DTT) dithiothreitol, 1% Triton X-100, 10 μg/mL RNase A (Sigma; R6513-250MG), 0.4 U/mL DNAse I (Invitrogen, I354Ba), with protease inhibitors (Roche; 11836170001)) at a concentration of 20,000 cells/μL on ice for 30 min. The suspension was centrifuged at 10,000 × g for 10 min at 4°C. Extracts were boiled for 5 min in the presence of 2× SDS sample buffer (20% glycerol, 10% DTT, 6% SDS, 500 mM Tris, pH 6.8).
Electrophoresis and protein gel blotting
Reaction mixtures were separated on an 8% SDS gel with a 4% stacking gel at 200 V for 35 min. Proteins were transferred to nitrocellulose with a wet electroblotter system (BioRad) for 17.5 h at 30 V and 90 mA. Subsequently, the membrane was blocked with either 5% low fat milk in TRIS buffered saline TBS-0.1% Tween-20 (TBST) or 2% BSA in TBS-0.1% Tween-20 (TBS-T), and incubated overnight with the indicated primary antibody as follows: anti-Chk1 (Santa Cruz; SC-8408; 1:200), anti-phospho ser345 Chk1 (Cell Signaling; 2348S; 1:2000), or anti-actin (Santa Cruz; sc-58673; 1:200). After washing, the membrane was incubated with alkaline phosphatase-coupled anti-mouse (Promega; S3721; 2500) or anti-rabbit antibodies (Millipore; AP132A; 1:2500). Western blots were performed at least 2 times.Citation50
Immunofluorescence microscopy
Cells were plated on glass coverslips for 48 h prior to treatment, then fixed in 3% formaldehyde for 20 min at room temperature. Fixation was quenched by addition of 50 mM NH4Cl in PBS, the cells were permeabilized for 5 min in 0.2% Triton X-100, and blocked with 3% BSA for 30 min. Cells were incubated with primary antibodies for 2 h at room temperature as follows: anti-histone γH2AX (Millipore; 05-636; 1:400), anti-cyclin B1 (Santa Cruz; SC-752; 1:100), or anti-lamin A/C (Santa Cruz; SC-6215; 1:150). After washing, cells were incubated with secondary antibodies at room temperature for 2 h as indicated: Alexa488 anti-mouse (Life Technologies; A11059; 1:400) for anti-histone γH2AX, Alexa488 anti-rabbit for cyclin B1 (Life Technologies; A11008; 1:400), and Alexa488 anti-goat (Life Technologies; A11059; 1:150) for anti-lamin A/C. Nuclei were stained with 300 nM 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 15 min prior to mounting. Cells were observed on an Olympus microscope operated by Infinity Capture Imaging software. Images were collected by the Infinity3 camera within the linear dynamic range. Images were prepared with Adobe Photoshop (CS3 10.0) software. Cells positive for histone γH2AX, cyclin B1, or lamin A/C were counted using ImageJ (IJ 1.46r) software. At least 200 cells were counted per experiments and experiments were repeats at least twice.
Scoring micronuclei
Micronucleated cells, individual micronuclei, and nucleoplasmic bridges (NPBs) were counted manually from DAPI stained cells. DAPI positive micronuclei were confirmed by additional staining methods: lamin A/C, histone γH2AX, or BrdU. At least 200 cells were counted for each experiment and experiments were repeated 3 times. Criteria for scoring micronuclei included the following: (1) separate (non-overlapping) extra-nuclear structures that were positive for DAPI; (2) intensity of DAPI staining in micronucleus did not exceed the main nucleus; (3) encased by nuclear envelope composed of the protein lamin A/C; and (4) a rounded shape.Citation17,34 DAPI staining intensity and shape of micronuclei were confirmed by visual observations.
3.8. 5-bromo-2′-deoxyuridine (BrdU) and 5-ethynyl-2′-deoyxuridine (Edu) assays
Cells were pre-labeled with 10 μM BrdU for 1 h or 5 μM EdU for 30 min at 37°C and then fixed with ethanol and permeabilized according to the manufacturers' conditions (5-bromo-2-deoxyuridine Labeling and Detection Kit 1; Roche; 11296736001) (Click EdU Alexa 488, Fisher Scientific). Cells were then stained with anti-BrdU for 1 h at 37°C (1:10) and then incubated with the anti-mouse-Ig-fluorescein secondary antibody for 1 h at 37°C (1:10).Citation15 Slides were then processed for immunofluorescence. BrdU labeling experiments were performed 3 times and EdU labeling experiments were performed twice.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Cancer Cell Laboratory for valuable discussions.
Funding
We thank Alberta Innovates-Technology Futures, Alberta Innovates Sustainability Fund and the University of Lethbridge for providing funding for this study.
Related Research Data
References
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/10.1038/nature11252
- Cancer Genome Atlas Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8; PMID:18772890; http://dx.doi.org/10.1038/nature07385
- Swift LH, Golsteyn RM. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int J Mole Sci 2014; 15:3403-31; http://dx.doi.org/10.3390/ijms15033403
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097-106; PMID:9323137; http://dx.doi.org/10.1016/S0092-8674(00)80375-X
- Swift LH, Golsteyn RM. The relationship between checkpoint adaptation and mitotic catastrophe in genomic changes in cancer cells. In: Kovalchuk I, Kovalchuk O, eds. Genome Stability: Elsevier, 2016
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886-95; PMID:10959836; http://dx.doi.org/10.1016/S0960-9822(00)00610-2
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-45; PMID:16327781; http://dx.doi.org/10.1038/ncb1337
- Rothblum-Oviatt CJ, Ryan CE, Piwnica-Worms H. 14-3-3 binding regulates catalytic activity of human Wee1 kinase. Cell Growth Differ 2001; 12:581-9; PMID:11751453
- Dalal SN, Schweitzer CM, Gan J, DeCaprio JA. Cytoplasmic localization of human cdc25C during interphase requires an intact 14-3-3 binding site. Mol Cell Biol 1999; 19:4465-79; PMID:10330186; http://dx.doi.org/10.1128/MCB.19.6.4465
- Swift LH, Golsteyn RM. Cytotoxic amounts of cisplatin induce either checkpoint adaptation or apoptosis in a concentration dependent manner in cancer cells. Biol Cell 2016; 108:1-22; PMID:26482322; http://dx.doi.org/10.1111/boc.201500056
- Syljuasen RG, Jensen S, Bartek J, Lukas J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res 2006; 66:10253-7; PMID:17079442; http://dx.doi.org/10.1158/0008-5472.CAN-06-2144
- Kubara PM, Kernéis S, Studeny A, Lanser BB, Meijer L, Golsteyn RM. Human cells enter mitosis with damaged DNA after treatment with pharmacological concentrations of genotoxic agents. Biochem J 2012; 446:373-81; PMID:22686412; http://dx.doi.org/10.1042/BJ20120385
- Tkacz-Stachowska K, Lund-Andersen C, Velissarou A, Myklebust JH, Stokke T, Syljuasen RG. The amount of DNA damage needed to activate the radiation-induced G2 checkpoint varies between single cells. Radiother Oncol 2011; 101:24-7; PMID:21722983; http://dx.doi.org/10.1016/j.radonc.2011.05.060
- Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta 2008; 1786:32-40; PMID:18549824
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012; 482:53-8; PMID:22258507; http://dx.doi.org/10.1038/nature10802
- Terradas M, Martin M, Tusell L, Genesca A. Genetic activities in micronuclei: is the DNA entrapped in micronuclei lost for the cell? Mutat Res 2010; 705:60-7; PMID:20307686; http://dx.doi.org/10.1016/j.mrrev.2010.03.004
- Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011; 26:125-32; PMID:21164193; http://dx.doi.org/10.1093/mutage/geq052
- Cornforth MN, Goodwin EH. Transmission of radiation-induced acentric chromosomal fragments to micronuclei in normal human fibroblasts. Radiat Res 1991; 126:210-7; PMID:2023991; http://dx.doi.org/10.2307/3577820
- Guerrero AA, Gamero MC, Trachana V, Futterer A, Pacios-Bras C, Diaz-Concha NP, Cigudosa JC, Martinez AC, van Wely KH. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci 2010; 107:4159-64; PMID:20142474; http://dx.doi.org/10.1073/pnas.0912143106
- Palmitelli M, de Campos-Nebel M, Gonzalez-Cid M. Progression of chromosomal damage induced by etoposide in G2 phase in a DNA-PKcs-deficient context. Chromosome Res 2015; 23:719-32; PMID:26152239; http://dx.doi.org/10.1007/s10577-015-9478-4
- Lim HK, Asharani PV, Hande MP. Enhanced genotoxicity of silver nanoparticles in DNA repair deficient mammalian cells. Front Genet 2012; 3:104; PMID:22707954; http://dx.doi.org/10.3389/fgene.2012.00104
- Li B, Senbabaoglu Y, Peng W, Yang ML, Xu J, Li JZ. Genomic estimates of aneuploid content in glioblastoma multiforme and improved classification. Clin Cancer Res 2012; 18:5595-605; PMID:22912392; http://dx.doi.org/10.1158/1078-0432.CCR-12-1427
- Akudugu JM, Bohm L. Micronuclei and apoptosis in glioma and neuroblastoma cell lines and role of other lesions in the reconstruction of cellular radiosensitivity. Radiat Environ Biophys 2001; 40:295-300; PMID:11820738; http://dx.doi.org/10.1007/s00411-001-0121-8
- Gurung RL, Lim SN, Khaw AK, Soon JF, Shenoy K, Mohamed Ali S, Jayapal M, Sethu S, Baskar R, Hande MP. Thymoquinone induces telomere shortening, DNA damage and apoptosis in human glioblastoma cells. PLoS One 2010; 5:e12124; PMID:20711342; http://dx.doi.org/10.1371/journal.pone.0012124
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148:59-71; PMID:22265402; http://dx.doi.org/10.1016/j.cell.2011.12.013
- Charlier C, Kintz P, Dubois N, Plomteux G. Fatal overdosage with cisplatin. J Anal Toxicol 2004; 28:138-40; PMID:15068570; http://dx.doi.org/10.1093/jat/28.2.138
- Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 2005; 5:231-7; PMID:15738985
- Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel E, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 1999; 59:3761-7; PMID:10446993
- Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 2013; 4:2130; PMID:23945651; http://dx.doi.org/10.1038/ncomms3130
- Wang XQ, Zhu YQ, Lui KS, Cai Q, Lu P, Poon RT. Aberrant Polo-like kinase 1-Cdc25A pathway in metastatic hepatocellular carcinoma. Clin Cancer Res 2008; 14:6813-20; PMID:18980975; http://dx.doi.org/10.1158/1078-0432.CCR-08-0626
- Thompson R, Meuth M, Woll P, Zhu Y, Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int J Oncol 2012; 40:194-202; PMID:21894433
- Mak JP, Man WY, Chow JP, Ma HT, Poon RY. Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget 2015; 6:21074-84; PMID:26025928; http://dx.doi.org/10.18632/oncotarget.4020
- Petsalaki E, Dandoulaki M, Morrice N, Zachos G. Chk1 protects against chromatin bridges by constitutively phosphorylating BLM serine 502 to inhibit BLM degradation. J Cell Sci 2014; 127:3902-8; PMID:25015292; http://dx.doi.org/10.1242/jcs.155176
- Okamoto A, Utani K, Shimizu N. DNA replication occurs in all lamina positive micronuclei, but never in lamina negative micronuclei. Mutagenesis 2012; 27:323-7; PMID:22086909; http://dx.doi.org/10.1093/mutage/ger082
- Krokan H, Wist E, Krokan RH. Aphidicolin inhibits DNA synthesis by DNA polymerase α and isolated nuclei by a similar mechanism. Nucleic Acids Res 1981; 9:4709-19; PMID:6795595; http://dx.doi.org/10.1093/nar/9.18.4709
- Shimura T, Martin MM, Torres MJ, Gu C, Pluth JM, DeBernardi MA, McDonald JS, Aladjem MI. DNA-PK is involved in repairing a transient surge of DNA breaks induced by deceleration of DNA replication. J Mol Biol 2007; 367:665-80; PMID:17280685; http://dx.doi.org/10.1016/j.jmb.2007.01.018
- Terzoudi GI, Karakosta M, Pantelias A, Hatzi VI, Karachristou I, Pantelias G. Stress induced by premature chromatin condensation triggers chromosome shattering and chromothripsis at DNA sites still replicating in micronuclei or multinucleate cells when primary nuclei enter mitosis. Mutat Res Genet Toxicol Environ Mutage 2015; 793:185-98; http://dx.doi.org/10.1016/j.mrgentox.2015.07.014
- Sabatinos SA, Ranatunga NS, Yuan JP, Green MD, Forsburg SL. Replication stress in early S phase generates apparent micronuclei and chromosome rearrangement in fission yeast. Mol Biol Cell 2015; 26:3439-50; PMID:26246602; http://dx.doi.org/10.1091/mbc.E15-05-0318
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013; 154:47-60; PMID:23827674; http://dx.doi.org/10.1016/j.cell.2013.06.007
- Leibowitz ML, Zhang CZ, Pellman D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu Rev Genet 2015; 49:183-211; PMID:26442848; http://dx.doi.org/10.1146/annurev-genet-120213-092228
- Deckbar D, Birraux J, Krempler A, Tchouandong L, Beucher A, Walker S, Stiff T, Jeggo P, Lobrich M. Chromosome breakage after G2 checkpoint release. J Cell Biol 2007; 176:749-55; PMID:17353355; http://dx.doi.org/10.1083/jcb.200612047
- Fink LS, Roell M, Caiazza E, Lerner C, Stamato T, Hrelia S, Lorenzini A, Sell C. 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging 2011; 3:836-45; PMID:21931182; http://dx.doi.org/10.18632/aging.100381
- Shimizu N, Itoh N, Utiyama H, Wahl GM. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol 1998; 140:1307-20; PMID:9508765; http://dx.doi.org/10.1083/jcb.140.6.1307
- Anderson CW, Allalunis-Turner MJ. Human TP53 from the malignant glioma-derived cell lines M059J and M059K has a cancer-associated mutation in exon 8. Radiat Res 2000; 154:473-6; PMID:11023613; http://dx.doi.org/10.1667/0033-7587(2000)154%5b0473:HTFTMG%5d2.0.CO;2
- Cahuzac N, Studény A, Marshall K, Versteege I, Wetenhall K, Pfeiffer B, Léonce S, Hickman JA, Pierré A, Golsteyn RM. An unusual DNA binding compound, S23906, induces mitotic catastrophe in cultured human cells. Cancer Lett 2010; 289:178-87; PMID:19758748; http://dx.doi.org/10.1016/j.canlet.2009.08.014
- Ferry G, Studény A, Bossard C, Kubara PM, Zeyer D, Renaud JP, Casara P, de Nanteuil G, Wierzbicki M, Pfeiffer M, et al. Characterization of novel Checkpoint kinase 1 inhibitors by in vitro assays and in human cancer cells treated with topoisomerase inhibitors. Life Sci 2011; 89:259-68; PMID:21736880; http://dx.doi.org/10.1016/j.lfs.2011.06.010
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/10.1126/science.282.5393.1497
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756-60; PMID:17051156; http://dx.doi.org/10.1038/nature05236
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55-63; PMID:6606682; http://dx.doi.org/10.1016/0022-1759(83)90303-4
- Lewis CW, Taylor RG, Kubara PM, Marshall K, Meijer L, Golsteyn RM. A western blot assay to measure cyclin dependent kinase activity in cells or in vitro without the use of radioisotopes. FEBS letters 2013; 587:3089-95; PMID:23954627; http://dx.doi.org/10.1016/j.febslet.2013.08.003