
ABSTRACT
Poly (ADP-ribose) polymerase (PARP) is a key molecule in the DNA damage response (DDR), which is a major target of both chemotherapies and radiotherapies. PARP inhibitors therefore comprise a promising class of anticancer therapeutics. In this study, we evaluated the efficacy of the PARP inhibitor olaparib, and also sought to identify the mechanism and predictive marker associated with olaparib sensitivity in head and neck cancer (HNC) cells. A total of 15 HNC cell lines, including AMC HNC cells, were tested. AMC-HN3 and HN4 exhibited stronger responses to olaparib. Among cisplatin-resistant cell lines, only AMC HN9-cisR cells were significantly suppressed by olaparib. We found that basal poly (ADP-ribose) (PAR) levels, but not PARP-1 levels, correlated with olaparib sensitivity. AMC-HN3 and HN4 cells exhibited higher basal levels of NF-κB that decreased significantly after olaparib treatment. In contrast, apoptotic proteins were intrinsically expressed in AMC-HN9-cisR cells. As interference with p53 expression led to NF-κB reactivation, we concluded that elevated basal PAR and NF-κB levels are predictive of olaparib responsiveness in HNC cells; in addition, olaparib inhibits HNC cells via PAR–p53–NF-κB interactions.
Introduction
The DNA damage response (DDR) is the primary target of radiotherapy and chemotherapeutic agents in the context of cancer treatment; however, this process is also at the forefront of anticancer therapy resistance mechanisms. Many drugs have been developed to inhibit repair mechanisms selectively in cancer cells and overcome subsequent treatment resistance. This relatively old concept remains the primary route of cancer treatment, despite daily developments in targeted therapeutics.Citation1
The DDR can be divided into 2 major categories: the homologous recombination (HR) pathway for double-strand break (DSB) repair, the base excision repair (BER), or nucleotide excision repair (NER) pathway for single-strand break (SSB) repair. Poly (ADP-ribose) polymerase (PARP) is a key molecule in SSB repair and plays opposing roles in cell fate decisions. In other words, PARP not only induces DNA repair to promote cell survival, but also induces cell extinction through diverse death-related signal transduction pathways induced by poly (ADP-ribose) (PAR), a process usually described as PARylation.Citation2-5 Other DDR pathway molecules, such as ataxia telangiectasia mutated (ATM), breast cancer gene (BRCA), and p53, also affect the functional directions of PARP and PAR; therefore, it is important to analyze and attempt to modulate the complex connections among those proteins in the context of cancer treatment.Citation6-8
PARP inhibitors currently hold the limelight in the field of cancer treatment, particularly as monotherapies or in combination with conventional chemotherapeutics for the treatment of BRCA 1/2-mutated, HR-deficient breast, ovarian, and prostate cancers.Citation9-13 Recently, a phase II clinical trial of olaparib, a well-known PARP inhibitor, combined with paclitaxel in patients with non-BRCA gene-mutated recurrent stomach cancer yielded positive results, suggesting the potential for the extended application of PARP inhibitors.Citation14 Few studies have evaluated PARP inhibitors in the context of head and neck cancers (HNCs), and these studies only compared responses according to different drug therapies or candidate gene mutations.Citation15,16
Based upon the above results, we hypothesized that PARP inhibitors might have a sufficient inhibitory effect on HNCs, which nearly all harbor wild-type (wt) BRCA. Additionally, we assumed that PARP inhibited cells would alter the responses of our cisplatin-resistant (cisR) HNC cells to cisplatin, in accordance with a report on the relationship between PARP hyperactivation and cisplatin resistance.Citation17 Although exciting activity has been seen with the use of PARP inhibitors in treating HR-deficient tumors, some patients still do not respond initially or develop acquired resistance with continued treatment. And resistance to PARP inhibitors during maintenance therapy is another recent challenge that has arisen in oncological practice. Explanations about the mechanism of PARP inhibitor resistance in BRCA 1/2 mutant tumors have been suggested, including HR pathway restoration, P-glycoprotein (PgP) overexpression, or p53 binding protein (53BP1) mutation.Citation18-21 We aimed, therefore, to discover a new resistance mechanism, along with a strategy to overcome such a mechanism, in HR-proficient tumors if we observed PARP inhibitor resistance in our HNC cells.
In this study, we primarily aimed to identify the effect of a single-agent PARP inhibitor on HNC cells. We further sought to investigate the possibility of overcoming cisplatin resistance using PARP inhibitors. Finally, we attempted to find a new mechanism or key signal molecule indicative of responsiveness to PARP inhibitors.
Results
Olaparib exerted selective inhibitory effects in some HNC cell lines
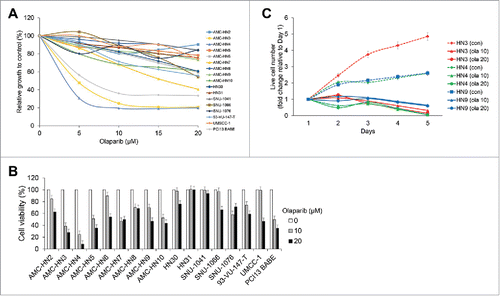
The cytotoxic effects of olaparib were evaluated in cultured human HNC cells. A few HNC cell lines, including HN3, HN4, and PCI13 BABE, demonstrated marked reductions in survival according to a MTT assay (). In a trypan blue exclusion assay, reductions in cell viability >50% were observed in HN5 and HN7 cells at a 10-μM concentration of olaparib (). Live cell numbers at the indicated time points, expressed as relative values to the cell counts on day 1, were compared, and the results suggested that the effects of olaparib on HNC cell growth were more likely cytostatic, rather than cytotoxic ().
Figure 1. Olaparib selectively inhibits some head and neck cancer (HNC) cell lines. The cytotoxic effects of olaparib were evaluated in cultured human HNC cells. (A) A few HNC cell lines, including AMC-HN3, -HN4, and PCI13 BABE, demonstrated marked decreases in survival in a MTT assay. (B) In a trypan blue exclusion assay, cell viability was reduced by more than 50% in AMC-HN5 and -HN7 cells at a 10-μM concentration of olaparib. (C) Live cell numbers at the indicated time points, expressed as relative values to the cell counts on day 1, were compared (con, control; ola 10 and 20, each olaparib 10 and 20 μM), revealing that the effects of olaparib on HNC cell growth were more likely cytostatic than cytotoxic. Error bars indicate standard errors.
Basal PAR expression levels correlated with olaparib responsiveness, even in cisplatin-resistant HNC cells
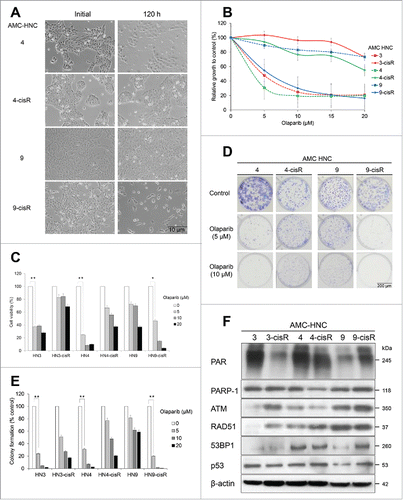
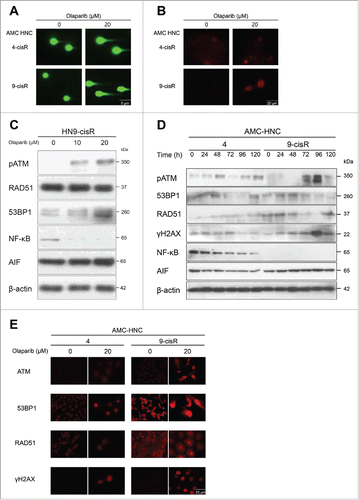
Following a previous report on the rescue of cisplatin resistance with PARP inhibitor, we applied olaparib to our cisplatin-resistant (cisR) HNC cells.Citation17 According to microscopic findings and MTT assays, we observed contrasting responses of HN-3/-4 and -9 cells and their corresponding cisR cells to olaparib treatment. Specifically, HN-3 and -4 cells were significantly inhibited by olaparib, whereas their corresponding cisR cells were not responsive. Inversely, cell viability was markedly decreased in HN9-cisR, but not HN9 cells (). These results were confirmed using trypan blue excursion and clonogenic assays (). We performed western blot analyses to determine the basal protein expression levels in each cell line as potential predictive markers of olaparib responsiveness. Relatively high levels of PAR expression appeared to correlate with olaparib sensitivity. However, neither PARP-1 overexpression nor decreased expression of HR-associated proteins, such as ATM, RAD51, and 53BP1, could explain olaparib responsiveness in the above-mentioned HNC cells ().
Figure 2. AMC-HN3, -HN4 and -HN9-cisR cells are sensitive to olaparib, and basal PAR expression levels correlate with olaparib responsiveness. (A) Cell morphology was evaluated using an inverted microscope after treatment with 10-μM olaparib for 120 h. (B) MTT assay according to changes in olaparib doses. (C) Trypan blue exclusion assay. (D and E) Clonogenic assay. (F) In western blot analyses, relatively high PAR expression levels appeared to be correlated with olaparib sensitivity. However, neither PARP-1 overexpression nor decreased expression HR-associated proteins could explain olaparib responsiveness in those HNC cells. The β-actin level was assessed as a loading control. * and ** denote p < 0.05 and p < 0.001, respectively. Error bars indicate standard errors.
Olaparib induced cell cycle changes and apoptotic cell death in HNC cells
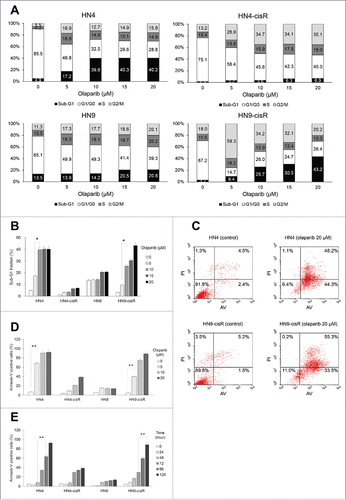
We wondered how olaparib could inhibit HR-proficient HNC cells in the absence of precedent DNA damage signals. Therefore, we performed cell cycle analyses after a 5-day olaparib treatment and observed significantly increased sub-G1 fractions in olaparib-sensitive HN4 and HN9-cisR cells ( and ). In AV-PI staining-based apoptosis assays, the AV-positive fractions among HN4 and HN9-cisR cells increased in a dose- and time-dependent manner, especially after 72 h ().
Figure 3. Olaparib led to cell cycle changes and apoptotic cell death. (A and B) According to a cell cycle analysis, the sub-G1 fractions were significantly increased in olaparib-sensitive AMC-HN4 and -HN9-cisR cells. (C-E) In Annexin-V FITC/PtdIns apoptosis assays, the Annexin-V-positive fractions in AMC-HN4 and -HN9-cisR cells were increased in a dose- and time-dependent manner, especially after 72 h. * and ** denote p < 0.05 and p < 0.001, respectively. Error bars indicate standard errors.
Olaparib induced HNC cell death via intrinsic apoptosis or another pathway
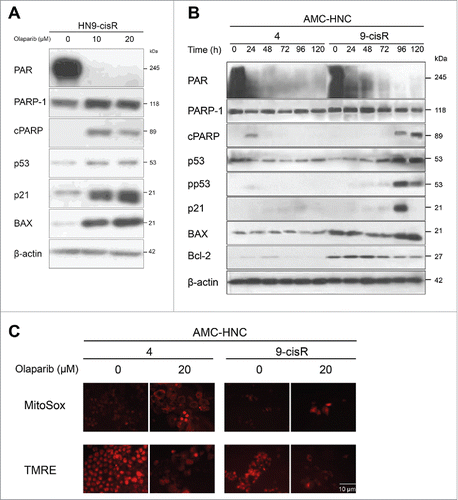
In HN9-cisR cells, a decrease in PAR and cleavage of PARP-1 with p21 and BAX activation were identified after a 72-h olaparib treatment (). Based upon the above results describing the cytostatic effect of olaparib with subsequent relatively late cell death, we also observed the expression of apoptotic proteins in HN4 and HN9-cisR cells for up to 120 h. We observed no definite expression of pp53, p21, or BAX after olaparib-induced PAR reduction in HN4 cells, unlikely in HN9-cisR cells (). Additional MitoSox and TMRE fluorescence tests were performed to confirm activation of the mitochondrial intrinsic apoptotic pathway; here, we observed weak but definite mitochondrial ROS production with membrane potential changes in both HN4 and HN9-cisR cells ().
Figure 4. Olaparib-induced cell death occurs via intrinsic apoptosis in AMC-HN9-cisR cells and through an undetermined process in AMC-HN4 cells. (A) Western blot analysis of AMC HN9-cisR cells according to changes in olaparib doses. Decreased PAR expression and PARP-1 cleavage, along with activated p21 and BAX, were identified after a 72-h olaparib treatment. (B) Western blot analyses of AMC-HN4 and -HN9-cisR cells according to the indicated time points after a 20-μM olaparib treatment. No definite expression of pp53, p21, or BAX was observed after olaparib-mediated PAR reduction in HN4 cells, unlikely in HN9-cisR cells. (C) Weak MitoSox and TMRE fluorescence were detected in both HN4 and HN9-cisR cells. Magnification: × 200.
Olaparib reduced the viability of HNC cells via suppression of NF-κB signaling
Because of the inconsistent findings concerning the olaparib-induced cell death mechanism in HN-3 and -4 cells, we investigated DDR pathways next. A comet assay and γH2AX IF assay were performed 72 h after olaparib treatment to identify DNA damage in both HN4-cisR (olaparib-resistant) and HN9-cisR cells (olaparib-sensitive). Although a relatively higher level of DNA damage was observed in HN9-cisR cells, olaparib also induced slight DNA damage in olaparib-resistant HN4-cisR cells (). To identify death mechanisms beyond the apoptotic cascades identified in HN-3 and -4 cells, we evaluated changes in the expression levels of HR pathway-related proteins in both HN4 and HN9-cisR cells after olaparib treatment. Additionally, we investigated PAR-associated molecular alterations by evaluating changes not only in AIF, a key molecule in parthanatos (PAR-induced cell death) but also in NF-κB, a well-known tumor-promoting signal transducer and target of PARylation.Citation3,22 Olaparib induced the activation of pATM, 53BP1, and γH2AX in a dose- and time-dependent manner in HN9-cisR cells (). However, in HN4 cells, no significant increase was observed in the expression of those DDR proteins, regardless of γH2AX accumulation. Intriguingly, NF-κB was more strongly expressed in HN4 cells compared to HN9-cisR cells, and the expression of this protein decreased significantly in a time-dependent manner after olaparib treatment (). The AIF level also appeared to have decreased in HN4 cells, although this finding was not consistently reproducible. The above results suggest that although olaparib could interfere with DDR in HNC cells, olaparib responsiveness might not be determined by the expression of HR-associated DDR proteins. We concluded that olaparib might induce cell death in HN-3 and -4 cells by suppressing PAR-associated NF-κB signaling.
Figure 5. Olaparib reduced the viability of AMC-HN4 cells via the suppression of NF-κB signaling. (A) A comet assay and (B) γH2AX immunofluorescence assay were performed 72 h after olaparib treatment to identify DNA damage. A relatively higher level of DNA damage was observed in HN9-cisR cells; however, olaparib also induced slight DNA damage in olaparib-resistant HN4-cisR cells. Magnification: × 100 (comet assay); × 400 (γH2AX). (C) Western blot analysis in HN9-cisR cells according to changes in olaparib doses. Olaparib induced pATM and 53BP1 activation in a dose-dependent manner in HN9-cisR cells. (D) Western blot analyses of HN4 and HN9-cisR cells according to the indicated time points after 20-μM olaparib treatment. NF-κB was more strongly expressed in HN4 cells, compared to HN9-cisR cells, and this expression decreased significantly in a time-dependent manner after olaparib treatment. (E) ATM, 53BP1, RAD51, and γH2AX immunofluorescence assays to detect DNA damage 72 h after olaparib treatment. Magnification: × 400.
The interaction between p53 and NF-κB might induce different cell death pathways in HNC cells after olaparib treatment
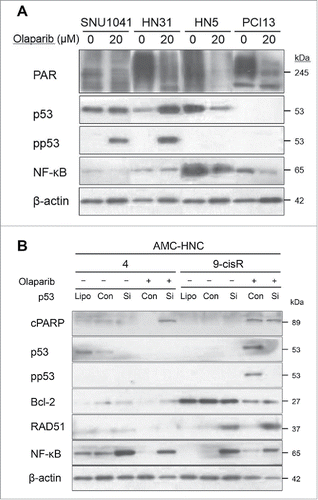
We sought to confirm a link between the PAR–NF-κB interaction and olaparib sensitivity in other HNC cell lines and selected the following representative cells according to the results of antecedent cell viability tests: olaparib-resistant, SNU-1041 and HN31; olaparib-sensitive, HN5 and PCI13 BABE. Similar to AMC HNC cells, the basal PAR expression levels in HN5 and PCI13 BABE cells were higher than those in SNU-1041 and HN31 cells, and more significant decreases in PAR expression were observed in the olaparib-sensitive cells after olaparib administration. We further reaffirmed these higher basal expression of NF-κB, along with a significant reduction after olaparib treatment in olaparib-sensitive HNC cells vs. resistant cells. Intriguingly, pp53 was only expressed in SNU-1041 and HN31 cells, and not in HN5 and PCI13 BABE cells (). We postulated that pp53 expression in olaparib-resistant cells might induce the cell survival pathway, in contrast to the apoptotic cascades observed in HN9-cisR cells. Based on those results, we expect that the p53–NF-κB interaction will be a crucial factor in determining olaparib responsiveness, and we investigated changes according to siRNA-mediated p53 modulation. Notably, p53 inhibition led to NF-κB overexpression in both naïve and olaparib-treated HN4 cells. In HN9-cisR cells, p53 inhibition plus olaparib treatment led to the disappearance of cleaved PARP; additionally, p53 inhibition also induced NF-κB activation (). We performed further experiment about the reciprocal changes on olaparib treatment after NF-κB modulation. That is, we investigated the effect of BAY 11-7089 (NF-κB inhibitor) and betulinic acid (NF-κB activator) in the olaparib-sensitive HN4 and HN9-cisR cells. In cytotoxicity assays, NF-κB activation with betulinic acid decreased olaparib-induced cell death in both HN4 and HN9-cisR cells. However, there was no significant changes on olaparib responsiveness after NF-κB inhibition with BAY 11-7089 in both HN4 and HN9-cisR cells (Fig. S1A). In Western blot analyses, we investigated the changes of PAR−p53−NF-κB levels with those NF-κB modulators. NF-κB activation with betulinic acid was observed in HN9-cisR cell and it might lead to a decreased olaparib response in HN9-cisR, but PAR decrement with p53 activation were consistently observed regardless of NF-κB regulations in HN9-cisR cell. However, in HN4 cell, there were inconstant changes of PAR−p53−NF-κB levels considering those cytotoxicity and NF changes and NF-κB modulations (Fig. S1B). To summarize the results above, p53 may interact reciprocally with NF-κB in both HN4 and HN9-cisR cells to regulate olaparib-induced cell death consequent to reduced PAR-mediated signaling through different pathways according to the basal NF-κB expression level.
Figure 6. The p53–NF-κB interaction might induce different cell death pathways in head and neck cancer (HNC) cells after olaparib treatment. (A) Western blot analyses of SNU-1041 and HN31 (olaparib-resistant) and HN5 and PCI13 BABE (olaparib-sensitive) cells at 72 h after olaparib treatment. The basal PAR expression levels were higher in HN5 and PCI13 BABE cells than in SNU-1041 and HN31 cells, and more significant decreases in PAR expression were observed in olaparib-sensitive cells after olaparib administration. Higher basal expression levels and more significant reductions in NF-κB were also observed in olaparib-sensitive HNC cells after olaparib treatment. (B) Changes in protein expression following siRNA-mediated p53 modulation. p53 inhibition caused NF-κB overexpression in both naïve and olaparib-treated HN4 cells. In HN9-cisR cells, p53 inhibition plus olaparib treatment led to the disappearance of cleaved PARP; additionally, p53 inhibition led to NF-κB activation.
Discussion
Most previous studies about the cancer therapeutic effects of PARP inhibitors were extremely skewed toward BRCA1/2-mutated HR-deficient cancers. In addition, even the few reports conducted in HR-proficient non-gynecologic solid tumors did not concretely evaluate the sole effects of PARP inhibitors, but rather evaluated the inhibitors in combination with DNA-damaging agents.Citation16,23,24 In this respect, our study is the first to identify the independent inhibitory effects of olaparib and reveal markers predictive of olaparib responsiveness, as well as the unique cell death mechanisms in various HNC cell lines. In addition, this study proposes the potential applicability of olaparib to cisplatin-resistant HNCs, which would be identified according to basal PAR overexpression, in contrast to a previous study of PARP-1 hyperactivation and cisplatin resistance in cancers.Citation17
p53 plays a pivotal role in DNA repair and recombination, and p53 propensity can decide cell fate.Citation7 There has been conflicting data regarding the effect of the p53 status of a cancer cell on olaparib responsiveness. One study reported that the effects of PARP inhibitors were increased when endogenous wt-p53 and the related HR activity were silenced in breast cancer cells.Citation25 In contrast, another study demonstrated that PARP inhibitor-induced DDR and cell death could be activated in both p53-dependent and -independent manners after delivering a DNA damage signal to HR-proficient cancers.Citation6 In the present study, p53 was activated after olaparib treatment mainly in olaparib-resistant HNC cells, suggesting that olaparib-mediated p53 activation in those cells might have led to DDR and the cell survival pathway. Conversely, p53 activation was also observed in olaparib-sensitive HN9-cisR cells along with strong PAR expression and relatively weak NF-κB expression. We can conclude that the role of p53 in the context of olaparib treatment is critically related to the basal expression of PAR and NF-κB in the cell.
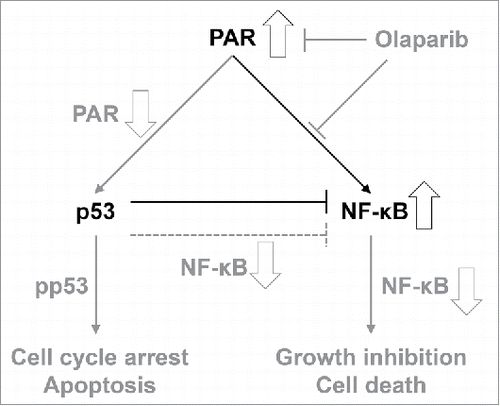
The reciprocal inhibition of p53 (tumor suppressor) and NF-κB (tumor enhancer) is well-known. We also observed NF-κB activation after p53 silencing in our study (). Many researchers have developed small molecule inhibitors with simultaneous p53 activating and NF-κB suppressing effects as cancer treatments.Citation26,27 In addition, PAR and PARP expression might lead to increased inflammation via upregulated NF-κB signaling, with subsequent tumor proliferation.Citation28,29 Therefore, the PAR–p53–NF-κB interaction would represent a promising cancer therapeutic target, and our study has identified some of the clues required to understand those connections. In summary, our study found that olaparib selectively killed PAR-overexpressing HNC cells by reducing PAR expression and subsequent PAR-associated signal transduction; this was recognized by p53, after which p53 might determine the HNC cell fate and death pathways according to the baseline level of NF-κB expression. In other words, HNC cells exhibiting basal NF-κB overexpression could be significantly affected by a decrease in PAR expression after olaparib treatment. On the other hand, reduced PAR expression might stimulate p53 and subsequent intrinsic apoptotic cascades in cells with low basal NF-κB but high PAR expression levels, such as HN9-cisR cells. We have depicted the mechanism underlying the effects of olaparib on HR-proficient HNC cells in .
Figure 7. Schematic mechanism by which olaparib affects homologous recombination (HR)-proficient head and neck cancer (HNC) cells. Olaparib-induced signal changes and subsequent results are indicated in gray.
In this study, we considered HNC cells without BRCA1/2 mutation to be HR-proficient cells; however, other HR pathway-related molecules and post-replication repair proteins should be considered to fully elucidate responsiveness to PARP inhibitors.Citation30 Furthermore, we cannot suggest specific indicators other than the basal PAR expression level that are predictive of olaparib sensitivity in HN-cisR cells, because of the inconsistent basal expression levels of predictable markers (e.g., PgP, Nrf-2, bcl-2) for drug resistance (Fig. S2). And we must have to prove the second aim of this study as the possibility of overcoming cisplatin resistance using PARP inhibitor, but we could not find any constant or significant combination effects between cisplatin and olaparib on both HNC cells and their paired cisplatin-resistant cells (Fig. S3). Finally, we must explain the inconsistent changes in olaparib sensitivities along with basal PAR expression levels in HN-3/-4 versus HN-9 cells as dependent on the acquisition of cisplatin resistance. In future experiments, we will attempt to resolve the aforementioned limitations of the current study and establish more specific indicators for olaparib treatment, with a particular focus on overcoming cisplatin resistance.
Conclusions
Our study clarified the efficacy of olaparib monotherapy against some HNC cells, including cisR cells. Basal PAR and NF-κB overexpression might be predictive markers of olaparib responsiveness in HR-proficient HNC cells. In addition, olaparib might suppress PAR and associated NF-κB signaling and induce cell death in olaparib-sensitive HNC cells, although this cannot be fully explained by an apoptotic mechanism. On the other hand, in HN9-cisR cells, which express relatively low levels of NF-κB, the olaparib-mediated decrease in PAR led to intrinsic apoptotic cell death via p53 activation.
Materials and methods
Cell culture and reagents
The following HNC cell lines were grown in a medium supplemented with 10% fetal bovine serum and subjected to cell viability tests: AMC-HN2, -HN3, -HN4, -HN6, -HN7, -HN8, -HN9, and -HN10 (grown in Eagle's minimum essential medium; Life Technologies™; Carlsbad, CA, USA); SNU-1041, -1066, and -1076, HN5, HN30, and HN31 (grown in Roswell Park Memorial Institute medium; Life Technologies™); and UMSCC-1, 93-VU-147-T, and PCI13 BABE (grown in Dulbecco's modified Eagle's medium; Life Technologies™). All cancer cell lines were authenticated by DNA (short-tandem-repeat) profiling, which was provided by the cell bank. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. Cisplatin-resistant AMC-HN3 (HN3-cisR), AMC-HN4 (HN4-cisR), and AMC-HN9 (HN9-cisR) cells were developed from the respective parental cisplatin-sensitive populations via continuous exposure to increasing concentrations of cisplatin.Citation31 Cisplatin resistance was evaluated in both resistant and parental cells using cell viability assays. Cells were exposed to different concentrations of olaparib for 72−120 h. Control cells were exposed to an equivalent amount of dimethyl sulfoxide (DMSO). Olaparib (AZD2281; Selleckchem, Houston, TX, USA) was dissolved in DMSO and reconstituted to 10 mM with H2O; aliquots of the drug were stored frozen at −20°C. Cisplatin (cis-platinum (II) diamine dichloride [CDDP], 5 mM in H2O; Sigma-Aldrich, St. Louis, MO, USA) was stored at 4°C until use.
Identification of BRCAness
The incidence of BRCA1/2 mutation is known to be extremely low in HNCs (approximately 3.6%).Citation32 However, we identified the BRCA gene proficiency (i.e., BRCAness) of HNC cells in our study using various methods. Specifically, we determined BRCAness in a genomically validated HNC sample that included UMSCC-1.Citation33 Additionally, somatic BRCA variants were screened in HNC samples from The Cancer Genome Atlas (TCGA) cohort with reference to supplementary data published by Heitmann et al. and the cBio cancer genomics portal.Citation15,32 Finally, we investigated BRCA gene variations in our AMC-HNC cell lines by detecting SNPs/mutations in these cells and compared the variations to previously described mutations in the COSMIC database.Citation34 Of the HNC cells in this study, we observed BRCA1/2 gene variations only in 93-VU-147-T cells.
Western blot analysis
For protein expression analysis, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, Waltham, MA, USA) at 4°C. Immunoblotting was subsequently performed using standard procedures. Briefly, a total of 50 μg of protein was resolved by 8%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose or polyvinylidene difluoride membranes, and probed with primary and secondary antibodies. The following primary antibodies were used: PAR (Enzo Life Sciences, Farmingdale, NY, USA); ATM, NF-κB p65 (NF-κB), p53, and RAD51 (Santa Cruz Biotechnology, Dallas, TX, USA); apoptosis-inducing factor (AIF), BAX, γH2AX, PARP-1 with cleaved PARP, p21WAF1/CIP1 (p21); BAY 11-7089 and betulinic acid (Tocris Bioscience, Bristol, UK), and phospho-p53-Ser15 (pp53) (Cell Signaling Technology, Danvers, MA, USA). β-actin (Sigma-Aldrich) was used as the loading control; all antibodies were used at dilutions ranging from 1:250 to 1:5,000.
Measurements of cell cycle and cell viability
Some previous studies involving PARP inhibitors recommended a longer time course for protein expression analyses involving cell viability tests (e.g., long-term colony formation assays rather than short-term MTT assays) because PARP inhibitor activity would be more prominent at later time points compared with other cytotoxic agents.Citation14,35 In cell cycle assays, cells were exposed to olaparib for 120 h. The treated cells were subsequently trypsinized, fixed overnight in ice-cold ethanol, and stained with propidium iodide (PtdIns; Sigma-Aldrich) for 30 min at 37°C. Cellular DNA contents were measured using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). For cell death assays, cells were cultured with olaparib or an equivalent amount of DMSO (vehicle control). After 120 h, cells were harvested, washed in ice-cold phosphate-buffered saline (PBS), and resuspended in binding buffer. Cells were then stained with Annexin V (AV)-fluorescein isothiocyanate (FITC) and PI using an AV apoptosis detection kit (BD Biosciences). All data was analyzed using Cell Quest software (BD Biosciences).
Cell viability was determined through trypan blue exclusion, MTT, and clonogenic assays. For trypan blue exclusion, cells were seeded at a density of 1 × 105 in 6-well plates, allowed to reach 60%–70% confluence, and then treated with olaparib for 120 h. The cells were then trypsinized, stained with 0.4% trypan blue (Life Technologies™), and counted using a hemocytometer. For MTT assays, cells were seeded at a density of 3–5 × 103 cells/well in 96-well plates, incubated overnight, and exposed to olaparib for 120 h. The cells were then exposed to the tetrazolium compound 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma-Aldrich) for 4 h; solubilization buffer was subsequently added for 2 h. The absorbance in each well was measured at 570 nm using a SpectraMax M2 microplate reader (Molecular Devices, Eugene, OR, USA). For clonogenic assays, cells were exposed to olaparib or DMSO for 48 h, then incubated in drug-free medium for 5–7 d. The wells were subsequently stained with 0.5% crystal violet solution, and the numbers of colonies were counted.
Immunofluorescence (IF) analysis
Cells were cultured on 96-well plates (BD Falcon, Franklin Lakes, NJ, USA). After 24 h, cells were treated with 20-μM olaparib for 72 h. The cells were then fixed with 4% PFA in PBS for 15 minutes, washed with PBS, permeabilized with 0.2% Triton-X100 for 5 minutes, and blocked with 3% bovine serum albumin (BSA) in phosphate buffered saline with Tween-20 (PBSt) for 15 minutes. After blocking, cells were incubated with primary antibodies for 1 h at room temperature, washed 3 times with PBSt, and incubated with secondary antibodies for 30 minutes at room temperature. Primary anti-ATM, -RAD51, -53BP1, and -γH2AX antibodies were used at dilutions of used 1:250 to 1:500, then samples were incubated for 1 h. Secondary antibodies were IF-labeled indirectly with rhodamine B (Sigma-Aldrich), and diamidino-2-phenylindole (DAPI) (Thermo Scientific) was used to stain DNA (nuclear staining).
Mitochondrial damage was evaluated using the fluorescent probes MitoSox™ (Invitrogen) and tetramethylrhodamine ethyl ester (TMRE) (Sigma-Aldrich) to detect mitochondrial superoxides and the membrane potential (Δψm), respectively. Images of labeled cells were acquired at excitation and emission wavelengths of 490 and 516 nm for MitoSox and 549 and 574 nm for TMRE (Zeiss LSM 510; Carl Zeiss, Inc., Oberkochen, Germany).
A comet assay was performed to semi-quantitatively measure DNA damage as a function of the comet tail length. Cells were mixed with low melting temperature agarose at 37°C. The precipitated and immobilized cells were subsequently loculated on a CometSlide™ to which olaparib and a lysis solution (for cell membrane and DNA histone removal) were applied. Finally, samples were subjected to alkaline electrophoresis, stained with intercalating dye and visualized via IF microscopy to reveal DNA breaks.
Transfection and infection
TP53 knockdown, AMC-HN9 cells, which express wt p53, were seeded onto 60-mm plates in a medium without antibiotics; after 18 h, the cells were transfected with 50 nmol/L small interfering RNA (siRNA) specific for human TP53 or a scrambled control siRNA (Santa Cruz Biotechnology). Transfections were conducted using Lipofectamine RNAi Max reagent (Life Technologies™). After 48 h, cells were exposed to olaparib for an additional 24-h period and subjected to protein expression analysis. To evaluate p53 expression, wt p53 was stably transfected into p53-null UMSCC-1 cells using a retroviral vector with a puromycin resistance cassette (Cell Biolabs Inc., San Diego, CA, USA). After 48 h, cells (60%–70% confluence) were infected overnight with virus-containing media supplemented with 4 μg/mL polybrene (EMD Millipore, Billerica, MA, USA). Selection was performed using 2 μg/mL puromycin (Sigma-Aldrich). Protein expression and knockdown were confirmed by western blotting with anti-p53 antibodies.
Statistical analysis
Experiments were conducted in triplicate and repeated independently at least twice. All statistical analyses were conducted using IBM SPSS Statistics Software (version 21.0; IBM Corporation, Armonk, NY, USA) and statistical significance was assessed using a one-tailed Student's t-test with P < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download Zip (1.9 MB)Funding
This study was supported by a grant (no. 2015R1A2A1A15054540) from the Basic Science Research Program through the National Research Foundation of Korea (NRF), Ministry of Science, ICT, and Future Planning, and a grant (no. HI15C2920) from the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Ministry of Health & Welfare, Seoul, Republic of Korea (J.L. Roh).
Related Research Data
References
- Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015; 15:166-80; PMID:25709118; http://dx.doi.org/10.1038/nrc3891
- Aredia F, Scovassi AI. Poly(ADP-ribose): a signaling molecule in different paradigms of cell death. Biochem Pharmacol 2014; 92:157-63; PMID:24976506; http://dx.doi.org/10.1016/j.bcp.2014.06.021
- Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 2014; 171:2000-16; PMID:24684389; http://dx.doi.org/10.1111/bph.12416
- Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer 2011; 105:1114-22; PMID:21989215; http://dx.doi.org/10.1038/bjc.2011.382
- Meng XW, Koh BD, Zhang JS, Flatten KS, Schneider PA, Billadeau DD, Hess AD, Smith BD, Karp JE, Kaufmann SH. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J Biol Chem 2014; 289:20543-58; PMID:24895135; http://dx.doi.org/10.1074/jbc.M114.549220
- Nguyen D, Zajac-Kaye M, Rubinstein L, Voeller D, Tomaszewski JE, Kummar S, Chen AP, Pommier Y, Doroshow JH, Yang SX. Poly(ADP-ribose) polymerase inhibition enhances p53-dependent and -independent DNA damage responses induced by DNA damaging agent. Cell Cycle 2011; 10:4074-82; PMID:22101337; http://dx.doi.org/10.4161/cc.10.23.18170
- Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 2005; 6:44-55; PMID:15688066; http://dx.doi.org/10.1038/nrm1546
- Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, Dean M, Tu L, Gilley D, Magliocco AM, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med 2012; 4:515-27; PMID:22416035; http://dx.doi.org/10.1002/emmm.201200229
- Balmana J, Tung NM, Isakoff SJ, Grana B, Ryan PD, Saura C, Lowe ES, Frewer P, Winer E, Baselga J, et al. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann Oncol 2014; 25:1656-63; PMID:24827126; http://dx.doi.org/10.1093/annonc/mdu187
- Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011; 12:852-61; PMID:21862407; http://dx.doi.org/10.1016/S1470-2045(11)70214-5
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015; 33:244-50; PMID:25366685; http://dx.doi.org/10.1200/JCO.2014.56.2728
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012; 366:1382-92; PMID:22452356; http://dx.doi.org/10.1056/NEJMoa1105535
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014; 15:852-61; PMID:24882434; http://dx.doi.org/10.1016/S1470-2045(14)70228-1
- Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, et al. Randomized, Double-Blind Phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol 2015; 33:3858-65; PMID:26282658; http://dx.doi.org/10.1200/JCO.2014.60.0320
- Heitmann J, Geeleher P, Zuo Z, Weichselbaum RR, Vokes EE, Fetscher S, Seiwert TY. Poly (ADP-ribose) polymerase inhibitor efficacy in head and neck cancer. Oral Oncol 2014; 50:825-31; PMID:25017803; http://dx.doi.org/10.1016/j.oraloncology.2014.06.004
- Nasuno T, Mimaki S, Okamoto M, Esumi H, Tsuchihara K. Effect of a poly(ADP-ribose) polymerase-1 inhibitor against esophageal squamous cell carcinoma cell lines. Cancer Sci 2014; 105:202-10; PMID:24219164; http://dx.doi.org/10.1111/cas.12322
- Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res 2013; 73:2271-80; PMID:23554447; http://dx.doi.org/10.1158/0008-5472.CAN-12-3000
- Ceccaldi R, O'Connor KW, Mouw KW, Li AY, Matulonis UA, D'Andrea AD, Konstantinopoulos PA. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res 2015; 75:628-34; PMID:25634215; http://dx.doi.org/10.1158/0008-5472.CAN-14-2593
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 2013; 3:68-81; PMID:23103855; http://dx.doi.org/10.1158/2159-8290.CD-12-0049
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008; 105:17079-84; PMID:18971340; http://dx.doi.org/10.1073/pnas.0806092105
- Wagner LM. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther 2015; 8:1931-9; PMID:26251615 http://dx.doi.org/10.2147/OTT.S69935
- Gilmore T, Gapuzan ME, Kalaitzidis D, Starczynowski D. Rel/NF-kappa B/I kappa B signal transduction in the generation and treatment of human cancer. Cancer Lett 2002; 181:1-9; PMID:12430173; http://dx.doi.org/10.1016/S0304-3835(01)00795-9
- Chow JP, Man WY, Mao M, Chen H, Cheung F, Nicholls J, Tsao SW, Li Lung M, Poon RY. PARP1 is overexpressed in nasopharyngeal carcinoma and its inhibition enhances radiotherapy. Mol Cancer Ther 2013; 12:2517-28; PMID:23979918; http://dx.doi.org/10.1158/1535-7163.MCT-13-0010
- Olaussen KA, Adam J, Vanhecke E, Vielh P, Pirker R, Friboulet L, Popper H, Robin A, Commo F, Thomale J, et al. PARP1 impact on DNA repair of platinum adducts: preclinical and clinical read-outs. Lung Cancer 2013; 80:216-22; PMID:23410825; http://dx.doi.org/10.1016/j.lungcan.2013.01.014
- Ireno IC, Wiehe RS, Stahl AI, Hampp S, Aydin S, Troester MA, Selivanova G, Wiesmuller L. Modulation of the poly (ADP-ribose) polymerase inhibitor response and DNA recombination in breast cancer cells by drugs affecting endogenous wild-type p53. Carcinogenesis 2014; 35:2273-82; PMID:25085902; http://dx.doi.org/10.1093/carcin/bgu160
- Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nat Rev Drug Discov 2008; 7:1031-40; PMID:19043452; http://dx.doi.org/10.1038/nrd2759
- Zhuang C, Miao Z, Wu Y, Guo Z, Li J, Yao J, Xing C, Sheng C, Zhang W. Double-edged swords as cancer therapeutics: novel, orally active, small molecules simultaneously inhibit p53-MDM2 interaction and the NF-kappaB pathway. J Med Chem 2014; 57:567-77; PMID:24428757; http://dx.doi.org/10.1021/jm401800k
- Swindall AF, Stanley JA, Yang ES. PARP-1: Friend or Foe of DNA damage and repair in Tumorigenesis? Cancers (Basel) 2013; 5:943-58; PMID:24202328; http://dx.doi.org/10.3390/cancers5030943
- Weaver AN, Yang ES. Beyond DNA Repair: Additional functions of PARP-1 in cancer. Front Oncol 2013; 3:290; PMID:24350055; http://dx.doi.org/10.3389/fonc.2013.00290
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012; 72:5588-99; PMID:23118055; http://dx.doi.org/10.1158/0008-5472.CAN-12-2753
- Nakamura M, Nakatani K, Uzawa K, Ono K, Uesugi H, Ogawara K, Shiiba M, Bukawa H, Yokoe H, Wada T, et al. Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R. Oncol Rep 2005; 14:1281-6; PMID:16211297
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/10.1158/2159-8290.CD-12-0095
- Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, Sturgis EM, Ow TJ, Lotan R, Carey TE, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res 2011; 17:7248-64; PMID:21868764; http://dx.doi.org/10.1158/1078-0432.CCR-11-0690
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet 2008; Chapter 10:Unit 10.1; PMID:18428421
- Faraoni I, Compagnone M, Lavorgna S, Angelini DF, Cencioni MT, Piras E, Panetta P, Ottone T, Dolci S, Venditti A, et al. BRCA1, PARP1 and gammaH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim Biophys Acta 2015; 1852:462-72; PMID:25483710; http://dx.doi.org/10.1016/j.bbadis.2014.12.001