
ABSTRACT
Mutations in the oligomerization domain of p53 are genetically linked to cancer susceptibility in Li-Fraumeni Syndrome. These mutations typically alter the oligomeric state of p53 and impair its transcriptional activity. Activation of p53 through tetramerization is required for its tumor suppressive function by inducing transcriptional programs that lead to cell fate decisions such as cell cycle arrest or apoptosis. How p53 chooses between these cell fate outcomes remains unclear. Here, we use 5 oligomeric variants of p53, including 2 novel p53 constructs, that yield either monomeric, dimeric or tetrameric forms of p53 and demonstrate that they induce distinct cellular activities and gene expression profiles that lead to different cell fate outcomes. We report that dimeric p53 variants are cytostatic and can arrest cell growth, but lack the ability to trigger apoptosis in p53-null cells. In contrast, p53 tetramers induce rapid apoptosis and cell growth arrest, while a monomeric variant is functionally inactive, supporting cell growth. In particular, the expression of pro-arrest CDKN1A and pro-apoptotic P53AIP1 genes are important cell fate determinants that are differentially regulated by the oligomeric state of p53. This study suggests that the most abundant oligomeric species of p53 present in resting cells, namely p53 dimers, neither promote cell growth or cell death and that shifting the oligomeric state equilibrium of p53 in cells toward monomers or tetramers is a key parameter in p53-based cell fate decisions.
Introduction
The assembly of p53 into oligomers represents a crucial factor for its tumor suppressive function.Citation1 This transcription factor binds cooperatively to DNA adjacent and within p53 responsive genes as a tetramer, often referred to as a dimer of dimers.Citation2 The activation of p53 through its oligomerization leads to the regulation of cell fate outcomes, such as growth arrest and apoptosis, by controlling the expression of target genes. These functions of p53 are essential for maintaining genomic integrity and preventing oncogenic transformation.Citation3,4 In addition, p53 status has been linked to tumor progression and response to therapy in multiple cancers.Citation5 However, the underlying mechanism linking the oligomerization state of p53 to gene expression and cell fate remains poorly understood.
Mutations in the TP53 gene encoding p53 are the most common alterations in human cancer.Citation6 The majority are missense mutations that occur in the central DNA binding domain (DBD; residues 94–293), and are often associated with an oncogenic gain-of-function.Citation7 Mutations in the C-terminal oligomerization domain (OD; residues 325–355) are less frequent and lead to a reduction or loss of transcriptional activity.Citation8,9 A number of germline TP53 mutations within the OD have been identified in association with Li-Fraumeni syndrome (LFS), a hereditary disorder causing the early onset of a wide spectrum of cancers.Citation10,11 The connection between mutations in the OD of TP53 and LFS highlights the importance of maintaining the oligomerization function of p53 as it relates to cellular homeostasis.
Oligomerization of p53 is a dynamic process that is regulated through protein-protein interactions and cellular p53 levels. Oligomerization can also influence the pattern of p53 post-translational modifications.Citation12,13 Multiple proteins that bind directly to p53, often to its OD, can modulate p53 oligomerization and stability. These protein interactions can either enhance tetramer formation (i.e. Atg7, MYBBPIA, BCCIP, RhoGAPs) or inhibit oligomerization to promote monomeric p53 species (i.e., RBEL1A, S100, ARC).Citation14-20 Structurally, the p53 OD consists of a β-strand followed by an α-helix.Citation21 Two p53 monomers interact through their ODs, which associate in an antiparallel manner by contact between β-strands and hydrophobic interactions involving α-helical residues to form a primary dimer. Two primary dimers then self-associate through an interface between α-helices to form a “dimer of dimers,” referred to as a p53 tetramer.Citation22 It was previously determined that the assembly of p53 molecules into dimers occurs co-translationally, on the polysome, while tetramerization takes place at a post-translational stage in solution.Citation23 Tetramer formation increases the avidity of p53 to bind to its DNA-binding element by 1000-fold relative to the monomeric p53 core domain, while a dimeric form of p53 (residues 94–360) created by the L344A mutation has been shown to interact with DNA with just a 6-fold lower affinity than tetrameric p53.Citation24 Dissociation constant measurements between oligomeric forms and the estimated basal concentration of p53 in cells (0.06 – 0.5 µM) suggest that under normal conditions, cellular p53 primarily exists as dimers.Citation25,26 Using a fluorescently-tagged p53 variant, it was also reported that within living cells, nearly 60% of the basal p53 cellular pool exists as dimers. A shift toward the tetrameric form (dimer of dimers) was observed under cellular stress with a concomitant decrease in p53 degradation.Citation27 Understanding the cellular activities of p53 in the context of a shift in equilibrium between oligomeric states within cells is critical to deciphering how p53 mediates cell proliferation and death, or leads to oncogenesis.
In order to delineate cellular functions associated with monomers, dimers and tetramers of p53 in the absence of shifting oligomeric states, we have generated p53 variants of fixed oligomeric form, and demonstrated that dimeric p53 species lack the ability of tetramers to trigger cell death or apoptosis, but retain growth arrest function. This finding indicates that alterations in the oligomerization status of p53 can influence the cell fate decisions between growth, arrest and apoptosis.
Results
Oligomeric variants of p53
Five p53 structures (wild-type (wt) p53 or variants with altered ODs yielding p53 monomers, dimers or tetramers) were generated to study their cellular functions. Variants include p53 L344P, which contains a single point mutation within the OD that abolishes oligomerization, forming only monomeric species ( and ). Two p53 variants were also constructed that assemble into functional dimers. First, p53 M340Q/L344R (MQLR), a p53 variant that is described as a “half-tetramer,”Citation28 and second, p53-2OD, which harbours 2 consecutive ODs that thermodynamically favor its self-association into a dimer of p53 molecules through a tetrameric arrangement of p53 ODs ().Citation29 This design is based on a previous report that a duplicated p53 OD (2 linked ODs) self-associates to form a dimer (dimer of monomers), in a geometry equivalent to the wt tetramer (dimer of dimers).Citation29 Lastly, 2 tetrameric structures were prepared, namely wt p53 and the tetrameric variant p53-CR (E343K, E346K and K351E), a construct that contains 3 mutated, spatially proximal residues within the OD region leading to a constructive charge reversal at the dimer-dimer interface. These mutations thus maintain the existing pattern of salt bridges at this interface () uniquely favoring their self-association into tetramers. Importantly, this mutated OD interface cannot pair with the wt p53 OD.Citation30
Figure 1. Oligomeric variants of p53. (A) Amino acid sequence of the modified OD from p53-2OD contains 2 ODs (residues 310–360 of wt p53) separated by a glycine-threonine flexible linker. Ribbon models of the predicted structure for 2 tandem ODs of p53-2OD as a dimer of monomers (blue and gray) were generated by homology modeling of the 1PES structure using SWISS-MODEL. Unstructured regions are colored black. (B) Amino acid sequence of the OD from p53-CR contains 3 charge-reversal mutations at positions 343, 346 and 351. A ribbon model of p53-CR illustrates the constructive charge interactions within the dimer-dimer interface of the p53 OD. (C) Glutaraldehyde crosslinking of p53 species isolated from H1299 cell lysates. Western blot detection of p53 monomers and oligomers resolved by SDS PAGE following lysate treatment with 0.025% glutaraldehyde. (D) Schematic drawing of wt p53 and oligomeric variants. (E) Cellular expression of p53 oligomeric variants in H1299 cells at 24 hours (odd numbered lanes) and 48 hours (even numbered lanes) as analyzed by Western blot of cell lysates using an anti-p53 antibody (DO-1).
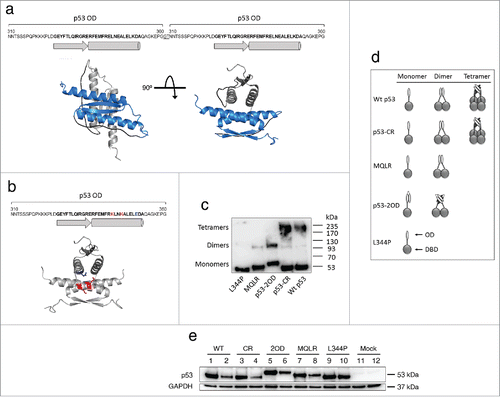
Glutaraldehyde crosslinking of p53 proteins from cell lysates of H1299 cells (p53-null) transiently expressing these p53 constructs confirmed that wt p53 and p53-CR do assemble into tetramers, MQLR and p53-2OD form dimers but not tetramers, while p53 L344P remains monomeric (). These p53 variants were shown to be equally expressed in H1299 cells 24 hours post-transfection as confirmed by western blot (). However, monomeric p53 L344P displayed a prolonged level of expression at 48 hours that can be attributed to its reported reduced ability to bind to MDM2.Citation11 Both dimeric species also have higher expression at 48 hours as compared to tetrameric p53s, indicating that the dimeric forms may be more stable. In addition, TP53 mRNA expression levels of all constructs were comparable to that of endogenous wt p53 expressed by human MCF-7 breast cancer cells, as measured by real-time quantitative reverse transcription PCR (qRT-PCR) (Fig. S1).
p53-mediated tumor suppression is controlled by its oligomeric state and requires the assembly of tetramers for rapid induction of apoptotic cell death
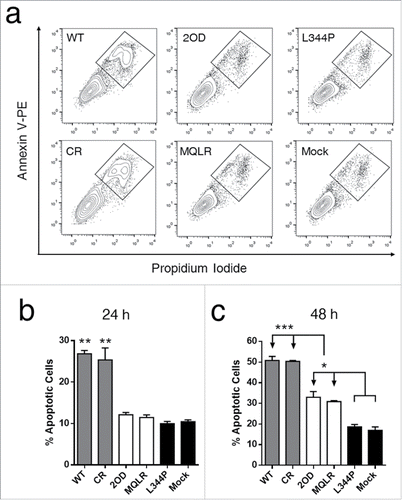
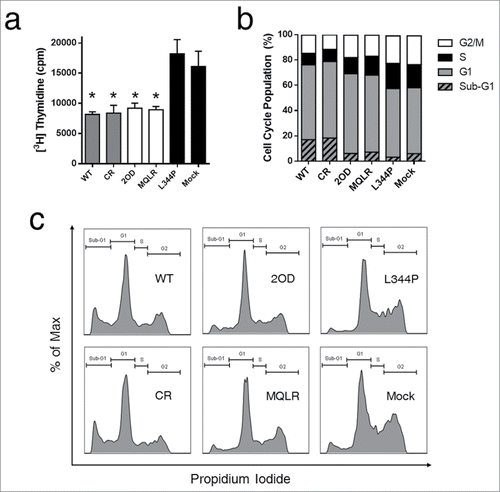
p53-dependent tumor suppression is primarily mediated through apoptosis and cell cycle arrest. Cell death and proliferation assays were performed in p53-null H1299 cells to establish the impact of controlling the oligomeric state of p53 on its cellular activities. First, tumor growth suppression induced by the cellular expression of p53 oligomeric variants was measured in real-time using a label-free cellular impedance monitoring device (). A distinct stepwise change in cell growth was observed between oligomeric variants, indicating that the oligomeric state of p53 directly correlates with cellular proliferation. As expected, tetrameric p53 species are potent tumor suppressors yielding the lowest level of cell proliferation while monomeric p53 was found to be functionally inactive resulting in the uncontrolled growth of p53 L344P-expressing p53-null H1299 cells (). In contrast, dimeric p53 exhibited approximately double the cellular expansion relative to p53 tetramers. Examination of cell viability by FACS analyses of propidium iodide (PI) stained H1299 cells revealed that only the expression of tetrameric p53 species induced cell death when compared to mock-transfected p53-null H1299 cells ( and ), where propidium iodide (PI) uptake is indicative of death. Another measure of cell viability is to assess the metabolic function of H1299 cells (NAD(P)H-dependent dehydrogenase activity) using a cell proliferation assay (MTS). This assay demonstrated that only tetrameric p53 species resulted in significant cell death (). A further flow cytometry-based analysis of apoptosis (Annexin V-PE/PI staining) confirmed that only tetramers induced rapid and efficient apoptotic cell death after 24 hours ( and ). However, a delayed and less robust apoptotic response was also observed after 48 hours of expression of p53 dimers ().
Figure 2. p53-mediated suppression of cell growth is related to its oligomeric state. (A) Cellular proliferation assay growth curve of H1299 cells expressing p53 oligomeric variants over 48 hours (mean ± SD) as measured by changes in cell surface impedance (xCELLigence RTCA DP analyzer). (B) Corresponding histogram depicting the area under the curve of H1299 cellular proliferation profiles (mean ± SD) after 48 hours.
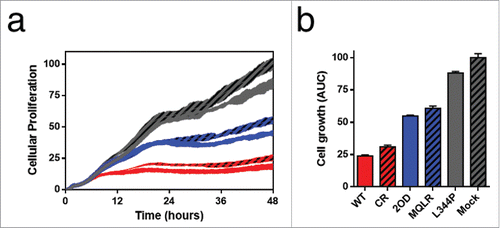
Figure 3. The tetrameric form of p53 promotes cell death. (a) Flow cytometric histograms of cell death profiles induced in H1299 cells expressing p53 variants. (b) Dead cell populations (PI stained) from cell death analysis (from panel a; mean ± SD (n = 3), ***p ≤ 0.001). (c) Measurement of cytotoxicity in H1299 cells expressing p53 variants using an MTS cell proliferation assay. Each bar represents the mean absorbance (490nm) ± SEM normalized to the mock transfection (n = 3), *p ≤ 0.05. NS, not significant.
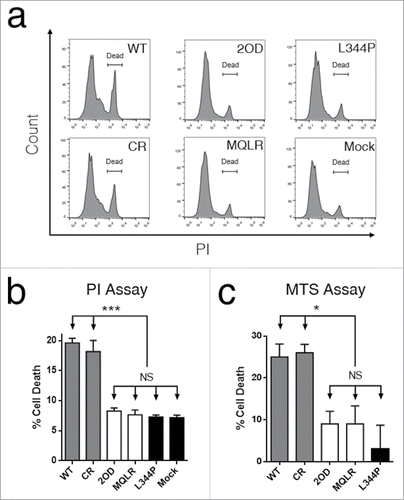
Figure 4. Only the expression of tetrameric p53 species induces rapid apoptosis in p53-null cells (Annexin V+, PI+ cells). (A) Representative flow cytometric plots of apoptosis in H1299 cells expressing p53 variants. (B) Apoptotic populations measured at 24 hours and (C) 48 hours. Each histogram bar represents the mean value ± SD (n = 3), *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
Dimeric p53 species exhibit growth arrest functions equivalent to p53 tetramers
Since the cellular expression of p53 dimers induced significant tumor suppression in p53-null H1299 cells () despite the relative lack of apoptotic cell death ( and ), we hypothesized that the tumor suppressor activity of p53 dimers is mediated predominantly through growth arrest. A subsequent analysis of DNA synthesis (thymidine uptake) demonstrated that dimers do inhibit DNA synthesis in H1299 cells at a level equivalent to that of tetrameric p53 species (). Cell cycle analyses also revealed that H1299 cells expressing p53 dimers and tetramers displayed larger G1 populations and smaller S and G2 populations, as compared to cells expressing monomeric p53 or mock-transfected cells; a distribution pattern indicative of cell cycle arrest ( and ). Furthermore, only p53 tetramer-expressing cells had a distribution profile with large sub-G1 peaks characteristic of fragmented nuclear DNA during apoptosis. Thus, while H1299 lung cancer cells transformed to express p53 dimers do not undergo efficient apoptosis, they are cytostatic, exhibiting growth inhibitory effects equivalent to cells expressing p53 tetramers; a cell cycle profile that accounts for their tumor suppressor activity.
Figure 5. Dimers of p53 inhibit DNA synthesis and cause cell cycle arrest in cells. (A) Thymidine incorporation assay in H1299 cells expressing p53 oligomeric variants. Each histogram bar represents the mean value ± SEM (n = 3). *p < 0 .05 compared to L344P. (B) Cell cycle analysis of H1299 cells expressing p53 oligomeric variants. (C) Representative flow cytometric plots of cell cycle analysis showing DNA content distributions.
Dimeric p53 species induce selective gene expression
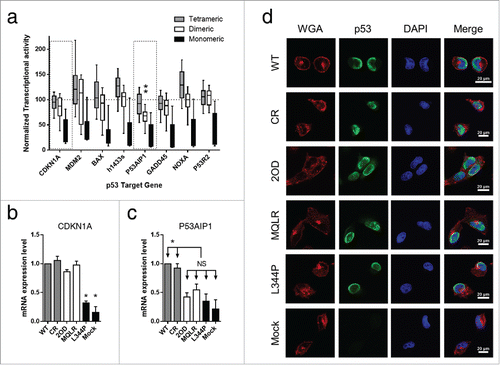
A comprehensive analysis of a missense mutation library in the OD of p53 generated by Kato et al. evaluated in the context of transactivation assays of p53 target genes performed in yeast, revealed that OD mutations alter p53 transcriptional activities as compared to wt p53.Citation8 To investigate the effect of oligomeric state on transcriptional activity, we performed a retrospective analysis of this data set, classifying OD mutations into categories that would predict monomeric, dimeric and tetrameric protein species (from crosslinking experiments).Citation9 The classification of OD mutations indicated a distinct step-wise correlation linking their transcriptional activity to their oligomeric status (). While it was observed that most dimeric p53 species display similar transcriptional activity as the tetrameric species, the induction of the p53 apoptosis-inducing protein 1 (P53AIP1) was the only gene for which the transcriptional activity was significantly reduced by OD mutations resulting in p53 dimers as compared to wt p53 and other tetrameric p53 mutants (; **p ≤ 0.0001). In agreement with this finding, gene expression analysis by qRT-PCR using RNA harvested from H1299 cells expressing p53 oligomeric variants produced similar results. Dimeric p53 species induced the mRNA expression of cyclin-dependent kinase inhibitor 1A (CDKN1A; p21, Cip1, Waf1) at levels equivalent to that observed for tetrameric p53 species, but had mRNA expression levels of P53AIP1 comparable to that of monomeric p53 and the mock transfection ( and ). An altered subcellular localization (i.e. interference with nuclear accumulation) may affect the ability of p53 to regulate the transcription of target genes. However, immunofluorescence confocal microscopy detecting WT p53 and all oligomeric variants expressed in H1299 cells indicates that all constructs localize to the nuclei. This finding suggests that the oligomeric state of p53 does not interfere with its appropriate nuclear localization (). Taken together, the selective loss of apoptosis-inducing P53AIP1 and the retention of CDKN1A gene expression support a functional outcome where the expression of p53 dimers leads to growth arrest but not cell death.
Figure 6. Retrospective analysis of transcriptional profiles of p53 oligomeric variants reveals that dimeric p53 mutants display selective gene expression. (A) Plot of transcriptional activity of selected p53 target genes performed in yeast using a missense mutation library in the OD of p53 (derived from data by Kato et al.).Citation8 Classification of 184 p53 mutants are categorized into monomeric, dimeric and tetrameric protein species as predicted from crosslinking experiments.Citation9 Box plots represent the 10–90th percentiles and are normalized to wt p53 activity (100%) represented by the dotted line (**p ≤ 0.0001). (B) mRNA expression analysis of CDKN1A and (C) P53AIP1 in p53-null H1299 cells expressing p53 oligomeric variants (mean value ± SEM, *p ≤ 0.05; relative expression was normalized to gapdh expression). (D) Representative subcellular localization of p53 oligomeric variants expressed in H1299 cells. Cells were counterstained with Alexa Fluor 594 conjugate of wheat germ agglutinin (WGA) to outline cells and DAPI to indicate the nuclei. p53 proteins were detected using anti-p53 mAb followed by a FITC-conjugated secondary Ab. All p53 variants demonstrate nuclear localization.
Discussion
The activation of p53 through its tetramerization is a key step in the cellular response to stressCitation1. The major functions of p53 are growth arrest and apoptosis, and the balance between these 2 cellular events can determine cell fate. The importance of p53 oligomerization and its role in tumorigenesis is exemplified by patients with LFS, where carriers of germline TP53 mutations have a high risk of developing a wide spectrum of early-onset cancers.Citation10,11 Defects in p53 oligomerization by the introduction of OD mutations have been shown to decrease the transcriptional activity of p53 in yeast and mammalian cells.Citation8,9,31 However, it is not fully understood how the oligomerization state of p53 impacts gene expression and the resulting cell fate outcome that leads to either tumor suppression or cellular transformation.
Dimers are the predominant form of p53 in the cellular pool based on the estimated basal concentration of p53 in cells, dissociation constant measurements between oligomeric forms, and observations in live cells using fluorescently-tagged p53 molecules.Citation25-27 A shift in equilibrium between oligomeric states can be controlled through multiple known protein-protein interactions, either by promoting the formation of tetramers or sequestering p53 in the monomeric state. Some interactions that modulate p53 oligomerization have been shown to influence p53 activity.Citation14,17,20 For example, the direct binding of apoptosis repressor with caspase recruitment domain (ARC) to the p53 OD inhibits both p53 tetramerization and p53-dependent apoptosis.Citation20 Alternatively, the Rho GTPase activation protein (RhoGAP) ArhGAP11A binds the p53 OD to stabilize tetramer formation, enhancing both cell cycle arrest and apoptosis activities. Intriguingly, RhoGAPs are frequently deleted or suppressed in cancers, indicating the importance of p53 tetramer formation in tumor suppression.Citation16 Despite these findings, a role for the most abundant cellular p53 species, namely dimers, has not been described.
Here, we demonstrate that the oligomerization status of p53 is key for cell fate decisions between growth, arrest or death. Although dimeric p53 species retained wt-like growth arrest functions through the inhibition of DNA synthesis and activation of cell cycle arrest (), they displayed only half of the activity of tetramers in a cell proliferation assay () that could be accounted for by the deficient induction of cell death compared to tetramers ( and ). In contrast, tetramers induced rapid apoptosis and cell growth arrest as potent tumor suppressors, while the monomeric L344P variant supported cell growth having no tumor suppressor function. Thus, the cellular function of p53 dimers is primarily mediated by growth arrest and a shift toward monomers or tetramers can steer the cell fate toward survival or death.
The defective cell death function of p53 dimers is reminiscent of previous reports of p53 mutations in its DBD that also display altered cell fate outcomes. For example, a single mutation in p53 (E177R) located at the interaction interface between DBDs results in the selective loss of apoptotic functions due to an altered quaternary structure of the p53 tetramer that was shown to hinder cooperative binding to DNA. Mutant p53E177R mice had abolished apoptotic functions but retained cell cycle control, and were nonetheless highly susceptible to cancer.Citation32 Similarly, mutant p53K117R mice were competent for cell cycle arrest but not apoptosis. In this case, the retention of non-apoptotic functions were sufficient to prevent early-onset spontaneous tumor formation.Citation33 In addition, the anti-apoptotic role of dominant negative mutant p53 R248Q is mediated through the suppression of P53AIP1 gene expression.Citation34 Alternately, an amino-terminal truncated p53 isoform (p47) demonstrated proficient apoptosis and prognosticates a better survival of cancer patients, although being insufficient for tumor suppression. This mutation is found in LFS patients. This mutant p53 has a compromised ability to induce genes involved in growth arrest, including CDKN1A, but was capable of inducing the pro-apoptotic gene P53AIP1.Citation35 In each case, the p53 variants displayed selective gene activation or altered gene expression patterns as compared to wt p53. Loss of function can also occur by mutating lysines 351 and 357 within the OD of p53 to glutamine residues; a p53 variant that forms tetramers when expressed in H1299 p53-null cells and impairs their cell cycle arrest function while retaining their ability to undergo cell death.Citation36 In contrast, the charge reversal mutation K351E in conjunction with E343K and E346K within the OD dimer-dimer interface of p53-CR forms tetramers in H1299 cells that retain all wt p53 functions.
The transcriptional targets of p53 directly control cellular outcomes.Citation37 Among these targets, CDKN1A induces cell cycle arrest, whereas P53AIP1 is pro-apoptotic and triggers the release of mitochondrial cytochrome c into the cytoplasm.Citation38 In studies of p53 expression dynamics and cell fate determination, it has been reported that cell cycle arrest and apoptosis are separately induced by the activation of CDKN1A and P53AIP1 gene expression, respectively.Citation39-41 Furthermore, the dependence of p53 on CDKN1A for cell cycle arrest and P53AIP1 for apoptosis has been established through inhibition assays and inducible-expression systems.Citation38,42-44 Our retrospective analysis of 184 p53 mutations in the OD, relating transcriptional activities to predicted oligomeric forms, revealed the selective loss of P53AIP1 gene expression among all dimeric p53 mutants, as compared to wt p53 and tetrameric p53 mutants ().Citation8,9 Moreover, dimeric p53 variants expressed in H1299 p53-null cells did not induce P53AIP1 gene expression but retained CDKN1A gene expression ( and ). As a result, we observed cytostasis due to abrogated apoptosis and intact cell growth arrest. In contrast, tetrameric species induced the expression of both genes with concomitant growth arrest and apoptotic functions. The monomeric p53 L344P had compromised transcriptional capability and possessed neither function. Thus, the expression of CDKN1A and P53AIP1 are, at least in part, important cell fate determinants, whose regulation is dependent on the oligomerization of p53. Taken together, these results indicate that different p53 oligomeric forms leads to different cell fates.
Materials and methods
p53 protein constructs
Point mutations were generated by site-directed mutagenesis of the wt human p53 cDNA inserted into a pcDNA3.1 mammalian expression vector. Oligonucleotide primers carrying the mutations of interest (Table S1) were used to amplify the TP53 gene by PCR and introduce the desired mutations. The amino acid sequence of p53-CR is available in Fig. S2A. The p53-2OD construct (amino acid sequence, Fig. S2B) was chemically synthesized and introduced into a pcDNA3.1 vector (GeneArt, Thermo Fisher Scientific).
Cell culture
The p53-null H1299 human non-small cell lung cancer cell line (ATCC No. CRL-5803) harbors a homozygous partial deletion of the TP53 gene leading to a loss of expression of the p53 protein. The cell line was maintained in Dulbecco's Modified Eagle Medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific), penicillin (100 units/mL) and streptomycin (100 µg/mL) (Thermo Fisher Scientific). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. Transient transfections of pcDNA3.1 p53 constructs were performed using Lipofectamine 2000 reagent following manufacturer's instructions (Thermo Fisher Scientific).
Western blot analysis
Total cell extracts were prepared in lysis buffer (25 mM HEPES pH 7.0, 250 mM NaCl, 0.1% Triton X-100) supplemented with Benzonase (1 units/mL; Novagen) and a cocktail of protease inhibitors (complete EDTA-free; Roche). Cell lysates were resolved by 4–12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad). p53 was detected using a mouse anti-human p53 monoclonal antibody (mAb) clone DO-1 (Santa Cruz Biotechnology). Equal loading and transfer to the membranes were verified by staining with anti-GAPDH mAb clone 6C5 (Thermo Fisher Scientific).
Oligomerization assay
Supernatants of lysed total cell extracts from transfected H1299 cells expressing p53 oligomeric variants were incubated in 0.025% glutaraldehyde (Sigma-Aldrich) prepared in phosphate buffered saline (PBS). Crosslinking reactions were left to proceed for 30 min at 4°C and reactions were ended with the addition of 100 mM glycine in PBS. Multimeric complexes were resolved by SDS-PAGE before transfer to nitrocellulose membrane (Bio-Rad) and protein gel blot analysis using anti-p53 mAb DO-1 to detect p53 protein complexes.
Immunofluorescence staining and confocal microscopy
H1299 cells expressing p53 oligomeric variants were grown on glass coverslips, fixed with 4% paraformaldehyde in PBS and permeabilized with ice-cold 100% methanol. Cells were stained with anti-p53 mAb DO-1 (1:500 dilution) followed by a fluorescein isothiocyanate (FITC)-conjugated secondary Ab (1:1000 dilution; Bethyl Laboratories) and counterstained with 1 µg/mL Alexa Fluor 594 conjugate of wheat germ agglutinin (WGA; Thermo Fisher Scientific) and 300 nM 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific). Images were collected with a Nikon A1 laser scanning confocal microscope at 40x magnification and analyzed using Nikon Imaging Software Elements Advanced Research Version 4.50.
Cellular proliferation assay
Cellular proliferation was measured in real-time using an xCELLigence RTCA DP label-free cellular impedance-monitoring device (ACEA Biosciences). Background impedance of cell culture media in each well was measured before adding cells. Transfected cells were suspended in fresh cell culture media and seeded at 2 × 104 cells per well into 16 well E-plates. The E-plate was incubated for 25 min at room temperature to allow the cells to settle in an evenly distributed pattern at the bottom of each well before inserting the E-plate into the RTCA DP instrument placed in a CO2 incubator. Changes in electrical impedance were recorded at 1 min intervals over a period of 48 h.
Cell death analysis
Dead cells were identified using propidium iodide (PI) staining following the manufacturer's instructions (Thermo Fisher Scientific). Briefly, cells were washed and resuspended in PBS at a concentration of 1 × 106 cells/mL. Next, 5 µL of PI was added to 100 µL of cell suspension and incubated for 15 min at room temperature in the dark. Flow cytometry was performed using a FACScalibur Cell Analyzer (BD Biosciences).
Cytotoxicity assay
A tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] was used to measure the viability of cells (Promega). H1299 cells expressing p53 variants were seeded at 5×103 cells per well into 96 well plates in quadruplicate. After 24 h, cells were incubated for 2 h with MTS solution, containing the electron-coupling reagent, phenazine methosulfate (Promega). Following the incubation with MTS solution, cell viability was assessed by measuring the absorbance (490 nm) of each well using a Synergy H1 microplate reade (BioTek).
Apoptosis assay
Apoptotic cells were identified using FITC-conjugated anti-annexin V antibody and PI dual stain following the manufacturer's instructions (BD PharMingen). Briefly, cells were washed with PBS and resuspended in 1 × binding buffer (BD PharMingen) at a concentration of 1 × 106 cells/mL. 100 µL of cell suspension was transferred to a conical tube and 5 µL each of annexin V-FITC and PI were added. Cells were incubated for 15 min at room temperature in the dark before analysis by flow cytometry using a FACScalibur Cell Analyzer. For each sample, 10,000 events were acquired in 3 independent experiments.
Thymidine incorporation
The incorporation of a radioactive nucleoside, 3H-thymidine, into new strands of chromosomal DNA during cell division was used to assess cellular proliferation. H1299 cells expressing p53 variants were seeded at 5 × 103 cells per well into 96 well plates in triplicate. After 24 h, cells were incubated for 4 h with 3H-thymidine (PerkinElmer) and washed twice in PBS before measuring radioactivity using a scintillation β-counter (TopCount, PerkinElmer).
Cell cycle analysis
PI staining of DNA was used to assess the cell cycle phase distribution. H1299 cells expressing p53 variants were harvested after 24 h expression and fixed in 5 mL ice-cold 70% ethanol. After 24 h of incubation at 4°C, cells were pelleted by centrifugation at 800 g, washed in PBS and treated with 50 µl of 100 µg/mL ribonuclease (Sigma-Aldrich). Cells were then incubated for 30 min with 200 µL of 50 µg/mL PI and analyzed by flow cytometry using a FACScalibur Cell Analyzer. For each sample, 5,000 events were acquired in 2 independent experiments.
Real-time quantitative reverse transcription PCR
Total RNA was extracted from H1299 cells after expressing p53 variants for 24 h using RNAeasy Mini kit (Qiagen). Synthesis of cDNA was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer's instructions. Real-time quantitative PCR was carried out on a CFX96 qPCR machine (Bio-Rad) and PCRs were performed in 96-well plates in 10 µL volumes using the following reaction conditions: 95°C for 3 min, followed by 40 cycles of 94°C for 15 sec and 60°C for 1 min. Oligonucleotide primers sequences for qPCR are listed in Table S2.
Statistical methods and data analysis
P values for experimental data were determined using the Student's t-test. GraphPad PRISM 6.0 software was used to plot all data and perform statistical analysis. Flow cytometry statistics were analyzed using FlowJo software version 10.0 (Tree Star). All experiments were performed independently at least 2 times.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Files
Download Zip (257 KB)Acknowledgments
We thank Dr. Nadeem Moghal for the generous use of equipment for qRT-PCR experiments and Dr. Cheryl Arrowsmith for the kind gift of the p53 variant MQLR.
Funding
The work was funded by a grant from the Canadian Institutes of Health Research to JG.
ORCID
Aaron Prodeus http://orcid.org/0000-0002-0719-3891
Related Research Data
References
- Chene P. The role of tetramerization in p53 function. Oncogene 2001; 20:2611-7; PMID:11420672; http://dx.doi.org/10.1038/sj.onc.1204373
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol Cell 2006; 22:741-53; PMID:16793544; http://dx.doi.org/10.1016/j.molcel.2006.05.015
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14:359-70; PMID:24739573; http://dx.doi.org/10.1038/nrc3711
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 1992; 356:215-21; PMID:1552940; http://dx.doi.org/10.1038/356215a0
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012; 26:1268-86; PMID:22713868; http://dx.doi.org/10.1101/gad.190678.112
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749-58; PMID:19776744; http://dx.doi.org/10.1038/nrc2723
- Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25:304-17; PMID:24651012; http://dx.doi.org/10.1016/j.ccr.2014.01.021
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA 2003; 100:8424-9; PMID:12826609; http://dx.doi.org/10.1073/pnas.1431692100
- Kawaguchi T, Kato S, Otsuka K, Watanabe G, Kumabe T, Tominaga T, Yoshimoto T, Ishioka C. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 2005; 24:6976-81; PMID:16007150; http://dx.doi.org/10.1038/sj.onc.1208839
- Malkin D. Li-Fraumeni syndrome. Genes Cancer 2011; 2:475-84; PMID:21779515; http://dx.doi.org/10.1177/1947601911413466
- Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene 1998; 17:643-9; PMID:9704930; http://dx.doi.org/10.1038/sj.onc.1201974
- Warnock LJ, Knox A, Mee TR, Raines SA, Milner J. Influence of tetramerisation on site-specific post-translational modifications of p53: a comparison of human and murine p53 tumor suppressor protein. Cancer Biol Ther 2008; 7:1481-9; PMID:18769132; http://dx.doi.org/10.4161/cbt.7.9.6473
- Itahana Y, Ke H, Zhang Y. p53 oligomerization is essential for its C-terminal lysine acetylation. J Biol Chem 2009; 284:5158-64; PMID:19106109; http://dx.doi.org/10.1074/jbc.M805696200
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012; 336:225-8; PMID:22499945; http://dx.doi.org/10.1126/science.1218395
- Ono W, Hayashi Y, Yokoyama W, Kuroda T, Kishimoto H, Ito I, Kimura K, Akaogi K, Waku T, Yanagisawa J. The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J Biol Chem 2014; 289:4928-40; PMID:24375404; http://dx.doi.org/10.1074/jbc.M113.474049
- Meng X, Yue J, Liu Z, Shen Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J Biol Chem 2007; 282:1570-1576; PMID:17135243; http://dx.doi.org/10.1074/jbc.M607520200
- Xu J, Zhou X, Wang J, Li Z, Kong X, Qian J, Hu Y, Fang JY. RhoGAPs attenuate cell proliferation by direct interaction with p53 tetramerization domain. Cell Rep 2013; 3:1526-38; PMID:23684608; http://dx.doi.org/10.1016/j.celrep.2013.04.017
- Lui K, Sheikh MS, Huang Y. Regulation of p53 oligomerization by Ras superfamily protein RBEL1A. Genes Cancer 2015; 6:307-16; PMID:26413214
- van Dieck J, Fernandez-Fernandez MR, Veprintsev DB, Fersht AR. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J Biol Chem 2009; 284:13804-11; PMID:19297317; http://dx.doi.org/10.1074/jbc.M901351200
- Foo RS, Nam YJ, Ostreicher MJ, Metzl MD, Whelan RS, Peng CF, Ashton AW, Fu W, Mani K, Chin SF, et al. Regulation of p53 tetramerization and nuclear export by ARC. Proc Natl Acad Sci USA 2007; 104:20826-31; PMID:18087040; http://dx.doi.org/10.1073/pnas.0710017104
- Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. Solution structure of the tetrameric minimum transforming domain of p53. Nat Struct Biol 1994; 1:877-90; PMID:7773777; http://dx.doi.org/10.1038/nsb1294-877
- Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 Å. Science 1995; 267:1498-502; PMID:7878469; http://dx.doi.org/10.1126/science.7878469
- Nicholls CD, McClure KG, Shields MA, Lee PWK. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. J Biol Chem 2002; 277:12937-45; PMID:11805092; http://dx.doi.org/10.1074/jbc.M108815200
- Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J Mol Biol 2004; 341:1145-59; PMID:15321712; http://dx.doi.org/10.1016/j.jmb.2004.06.071
- Ma L, Wagner J, Rice JJ, Hu W, Levine AJ, Stolovitzky GA. A plausible model for the digital response of p53 to DNA damage. Proc Natl Acad Sci USA 2005; 102:14266-71; PMID:16186499; http://dx.doi.org/10.1073/pnas.0501352102
- Rajagopalan S, Huang F, Fersht AR. Single-molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res 2011; 29:2294-303; http://dx.doi.org/10.1093/nar/gkq800
- Gaglia G, Guan Y, Shah JV, Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci USA 2013; 110:15497-501; PMID:24006363; http://dx.doi.org/10.1073/pnas.1311126110
- Davison TS, Nie X, Ma W, Lin Y, Kay C, Benchimol S, Arrowsmith CH. Structure and functionality of a designed p53 dimer. J Mol Biol 2001; 307:605-17; PMID:11254385; http://dx.doi.org/10.1006/jmbi.2001.4450
- Poon GM, Brokx RD, Sung M, Gariépy J. Tandem dimerization of the human p53 tetramerization domain stabilizes a primary dimer intermediate and dramatically enhances its oligomeric stability. J Mol Biol 2007; 365:1217-31; PMID:17113101; http://dx.doi.org/10.1016/j.jmb.2006.10.051
- Brokx RD, Bolewska-Pedyczak E, Gariépy J. A stable human p53 heterotetramer based on constructive charge interactions within the tetramerization domain. J Biol Chem 2003; 278:2327-32; PMID:12433927; http://dx.doi.org/10.1074/jbc.M208528200
- Lang V, Pallara C, Zabala A, Lobato-Gil S, Lopitz-Otsoa F, Farrás R, Hjerpe R, Torres-Ramos M, Zabaleta L, Blattner C, et al. Tetramerization-defects of p53 result in aberrant ubiquitylation and transcriptional activity. Mol Oncol 2014; 8:1026-42; PMID:24816189; http://dx.doi.org/10.1016/j.molonc.2014.04.002
- Timofeev O, Schlereth K, Wanzel M, Braun A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsässer HP, Stiewe T. p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep 2013; 30:1512-25; http://dx.doi.org/10.1016/j.celrep.2013.04.008
- Li T, Kon N, Tan M, Ludwig T, Zhao Y, Baer R, Wei G. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269-83; PMID:22682249; http://dx.doi.org/10.1016/j.cell.2012.04.026
- Igarashi H, Hirano H, Yahagi A, Saika T, Ishihara K. Anti-apoptotic roles for the mutant p53R248Q through suppression of p53-regulated apoptosis-inducing protein 1 in the RA-derived fibroblast-like synoviocyte cell line MH7A. Clin Immunol 2014; 150:12-21; PMID:24316591; http://dx.doi.org/10.1016/j.clim.2013.10.013
- Phang BH, Othman R, Bougeard G, Chia RH, Frebourg T, Tang CL, Cheah PY, Sabapathy K. Amino-terminal p53 mutations lead to expression of apoptosis proficient p47 and prognosticate better survival, but predispose to tumorigenesis. Proc Natl Acad Sci USA 2015; 112:E6349-58; PMID:26578795; http://dx.doi.org/10.1073/pnas.1510043112
- Beckerman R, Yoh K, Mattia-Sansobrino M, Zupnick A, Laptenko O, Karni-Schmidt O, Ahn J, Byeon I, Keezer S, Prives C. Lysines in the tetramerization domain of p53 selectively modulate G1 arrest. Cell Cycle 2016; 15:1425-38; PMID:27210019; http://dx.doi.org/10.1080/15384101.2016.1170270
- Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217-36; PMID:24577402; http://dx.doi.org/10.1038/nrd4288
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tikino T, Nakamaru Y, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by ser-46 phosphorylated p53. Cell 2000; 102:849-62; PMID:11030628; http://dx.doi.org/10.1016/S0092-8674(00)00073-8
- Zhang XP, Liu F, Cheng Z, Wang W. Cell fate decision mediated by p53 pulses. Proc Natl Acad Sci USA 2009; 106:12245-50; PMID:19617533; http://dx.doi.org/10.1073/pnas.0813088106
- Chen X, Chen J, Gan S, Guan G, Zhou Y, Ouyang Q, Shi J. DNA damage strength modulates a bimodal switch of p53 dynamics for cell-fate control. BMC Biol 2013; 11:73; PMID:23800173; http://dx.doi.org/10.1186/1741-7007-11-73
- Zhang XP, Liu F, Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc Natl Acad Sci USA 2011; 108:8990-5; PMID:21576488; http://dx.doi.org/10.1073/pnas.1100600108
- Matsuda K, Yoshida K, Taya Y, Nakamura K, Nakamura Y, Arakawa H. p53AIP1 regulates the mitochondrial apoptotic pathway. Cancer Res 2002; 62:2883-9; PMID:12019168
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817-25; PMID:8242752; http://dx.doi.org/10.1016/0092-8674(93)90500-P
- Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187-90; PMID:7585571