
ABSTRACT
The usage of metabolic intermediates as substrates for chromatin-modifying enzymes provides a direct link between the metabolic state of the cell and epigenetics. Because this metabolism-epigenetics axis can regulate not only normal but also diseased states, it is reasonable to suggest that manipulating the epigenome via metabolic interventions may improve the clinical manifestation of age-related diseases including cancer. Using a model of BRCA1 haploinsufficiency-driven accelerated geroncogenesis, we recently tested the hypothesis that: 1.) metabolic rewiring of the mitochondrial biosynthetic nodes that overproduce epigenetic metabolites such as acetyl-CoA should promote cancer-related acetylation of histone H3 marks; 2.) metformin-induced restriction of mitochondrial biosynthetic capacity should manifest in the epigenetic regulation of histone acetylation. We now provide one of the first examples of how metformin-driven metabolic shifts such as reduction of the 2-carbon epigenetic substrate acetyl-CoA is sufficient to correct specific histone H3 acetylation marks in cancer-prone human epithelial cells. The ability of metformin to regulate mitonuclear communication and modulate the epigenetic landscape in genomically unstable pre-cancerous cells might guide the development of new metabolo-epigenetic strategies for cancer prevention and therapy.
While metabolic rewiring of cancer cells can be a direct consequence of the concerted action of oncogenes and tumor suppressor genes that drive aberrant growth of cancer tissues, altered metabolism might also play a primary, causative role in oncogenesis. Accordingly, metabolic reprogramming is increasingly appreciated as a candidate hallmark of tumorigenesis.Citation1-5 In a recently proposed metabolism-centered model of carcinogenesis, known as geroncogenesisCitation2 the possibility of acquiring genetic aberrations increases when aging itself operates as a driver of cancer-like metabolic landscapes. Tumor formation might therefore depend not only on accumulating mutations in the genome, but also on the decline in the homeostasis or metabolic health that naturally occurs in aging cells. Indeed, after many years of being subordinated to the nucleus as the commander-in-chief, able to initiate biological events, the capacity of metabolism to independently dictate cellular actions is increasingly recognized among most cell biologists. Accordingly, the research community now acknowledges that aging-driven alteration of metabolic homeostasis might be sufficient to favor an imbalance in self-amplifying metabolic landscapes that ultimately manifest as aging-driven diseases, including cancer.Citation6
Metabolic programs: From anabolic supporters of genomic instability to epigenetic regulators of carcinogenesis
The metabolic health of our cells might be a key determinant factor pushing the risk balance or cancer risk-meter toward health or the risk of cancer.Citation1,2 On the one hand, genomic instability, a characteristic of almost all human cancers, can be perpetuating or limiting, as it comes at the cost of meeting a minimum set of metabolic requirements. Genomic instability can occur prior to or as a consequence of malignant transformation and forces cells to undergo metabolic adaptation to promote their survival and growth. The competitive advantages provided by some bioenergetic programs associated with resistance to cell death and biosynthetic programs capable of providing building blocks for cell growth and mitogenesis might expedite significant changes in metabolic functioning. The establishment of such aberrant metabolic states compatible and permissive with increased genomic instability would allow the accumulation of additional genetic and epigenetic alterations during carcinogenesis. Aging-related chronic activation of anabolic metabolism supporting cell growth and proliferation could favor a metabolic scenario for later oncogenic stimuli to complete the journey from non-cancerous to cancerous states.Citation2,6
On the other hand, the so-called oncometabolites and gerometabolites have been defined as small-molecule components (or enantiomers) of normal metabolism whose accumulation or depletion, respectively, causes a significant impact on the 2 primary epigenetic codes, histone modification and DNA methylation, establishing an epigenetic milieu capable of initiating carcinogenesis and/or driving aging.Citation3,4,7-11 Although it remains intriguing how aging-related changes in cellular metabolism might control the layers of epigenetic instructions that influence cell fate without altering the primary DNA sequence, it should be acknowledged that the multiple processes involved in the alteration of the chromatin state, including post-translational modifications of histone proteins, incorporation of specific histone variants, methylation of DNA and ATP-dependent chromatin remodeling, are likely the pivotal molecular bridges that mediate the direct communication between the metabolic and the chromatin state, and the consequent epigenetic targeting of cell fate regulatory genes.Citation12-20 Indeed, because the usage of metabolic intermediates as substrates for chromatin-modifying enzymes provides a direct link between the metabolic state of the cell and epigenetics, one can envision that the spatio-temporal distribution of the levels and types of specific metabolites might operate as key cancer-related molecular events, rendering a cell susceptible to the epigenetic rewiring required for the acquisition of an aberrant cancer cell state and, concurrently, of refractoriness to normal differentiation.
Metabolo-epigenetic clocks in aging-driven cancer: Understanding the link to delineate interventions
Given that the metabolism-epigenetics axis can regulate not only normal but also diseased states, it is reasonable to suggest that manipulating the epigenome through metabolic intervention should improve the manifestation of age-related diseases including cancer. In other words, if aging-driven metabolic reprogramming in normal tissues can significantly alter the endogenous metabolic production of bona fide etiological determinants of cancer, such as oncometabolites and gerometabolites, this undesirable trade-off between aging and cellular metabolism might pave the way for the epigenetic initiation of carcinogenesis in a strictly metabolic-dependent manner. Aging-driven, tissue-specific alterations in critical metabolic factors for de/methylation, de/acetylation, or de/phosphorylation dynamics in the nuclear epigenome (e.g., acetyl-CoA, α-ketoglutarate, NAD+, FAD, ATP, or S-adenosylmethionine) might induce faulty epigenetic reprogramming capable of redirecting cell-specific differentiation of adult cells into cancer-prone cellular states.Citation2
Specific patterns of metabolites or metabolic signatures can poise cells with chromatin states competent for rapid dedifferentiation–a hallmark of malignancy–while concomitantly allowing the acceleration of the rate at which a bioenergetic/biosynthetic threshold would permit the survival and expansion of (epi)genomically-altered cells. In this geroncogenic scenario, aging-driven metabolic changes might operate, in a subtle but significant manner, as true oncogenic events without markedly changing cellular phenotypes until they are qualitatively or quantitatively sufficient to be selectively advantageous in the tumor microenvironment. Unfortunately, a robust experimental framework to model and verify the notion that age-related metabolic changes could per se underlie tumorigenesis is lacking. Moreover, our ability to decipher how epigenetic traits are affected by metabolic signals remains limited despite the ever-growing evidence that the prevalent landscape of epigenetic modifications in any cell type might be viewed as a snapshot of its metabolic status. Indeed, beyond the well-characterized examples of how accumulation of some competitive oncometabolites due to mutations in the metabolic enzymes SDH, FH, and IDH can drive phenotypic changes through epigenetic mechanisms,Citation21-29 very few studies have assessed whether more transient changes in metabolism without the involvement of oncogenic mutations in Krebs cycle enzymes are also capable of exerting epigenetic effects.Citation19,30-32
BRCA1-driven accelerated geroncogenesis: A framework for modeling and evaluating aging-driven alterations in the metabolism-epigenetic axis
To hasten the assessment of how the metabolic milieu impacts on epigenetics to advance/delay the “metabolo-epigenetic clock” that controls aging-driven diseases such as cancer, we recently envisioned that, similar to the use of human progeroid syndromes (e.g., Hutchinson-Gilford progeria and Werner's syndrome) as accelerated aging models recapitulating some features of normal aging, it might be possible to employ accelerated models of aging-associated cancer to rapidly and accurately dissect and therapeutically test the metabolic requirements for the epigenetic traits that underlie aging-driven cancer risk. Based on the geroncogenesis hypothesis that postulates that aberrations in cellular metabolism naturally occurring with aging drives a field effect, predisposing normal tissues for cancer development, we proposed that mutations in cancer susceptibility genes such as BRCA1 might trigger accelerated geroncogenesis in breast epitheliaCitation33 BRCA1-driven accelerated geroncogenesisCitation33 establishes that: a.) The reprogramming of bioenergetic/biosynthetic metabolic programs induced by BRCA1 haploinsufficiency should result in a significant reduction in the time required for breast epithelial cells to fully phenocopy a cancer-like anabolic metabolism; b.) The alteration of metabolic co-factors closely associated with chromatin-modifying processes of active de/methylation, de/acetylation, and de/phosphorylation induced by BRCA1 haploinsufficiency might impart faulty epigenetic programs, which might drastically alter cell-specific differentiation before the development of disease.
Acetyl-coA: A proof-of-concept epigenetic metabolite to test the ability of metformin to modify histone acetylation
Although the use of pharmacologicals that directly regulate the activity of epigenetic enzymes is a well-established technique to modify the epigenetic landscape and cell phenotypes, we envisioned that the sole alteration of the availability of key metabolites used by chromatin-modifying enzymes could be an alternate approach to modulate the epigenetics of cancer-prone aging cells.Citation33 As such, alterations in cellular metabolism such as the restriction of mitochondrial biosynthetic capacity imposed by the biguanide metformin should manifest in global epigenetic effects. In line with recent studies proposing a “substrate limitation” model of metformin action,Citation34,35 we recently revealed that the anti-diabetic/anti-aging drug metforminCitation36-39 impedes the production of several mitochondrial-dependent biosynthetic intermediates by reducing the anaplerotic flux of glucose, glutamine, and likely branched-chain amino acids,Citation40 into the tricarboxylic acid (TCA) cycle, leading to the depletion of acetyl-CoA and malonyl-CoA required for de novo lipid biosynthesis and inhibition of mTOR-driven protein synthesis in anabolism-addicted BRCA1 haploinsufficient cells.Citation41
To mark histones post-translationally, chromatin modifiers use metabolic intermediates such as acetyl-CoA, the key cofactor used by histone acetyltransferases (HATs). Hence, histone modifications may reflect the metabolic status of cells and be influenced by the concentrations of epigenetic metabolites such as acetyl-CoA. Accordingly, histone acetylation is intimately associated with the cellular acetyl-CoA pool in response to metabolic state as it depends on intermediary metabolism for supplying acetyl-CoA in the nucleocytosolic compartment.Citation20,42-46 The fact that the mutation of a single BRCA1 allele dramatically alters the metabolomic signature of normal-like breast epithelial cells to induce a significant >20-fold augmentation of acetyl-CoACitation41 supports the notion that this model of BRCA1 haploinsufficiency might be relevant to directly explore:
whether histone acetylations are influenced by these differing concentrations of acetyl-CoA between normal-like and pre-diseased, cancer-prone epithelial cells, and
whether restricting the mitochondrial production of acetyl-CoA with metformin is sufficient to correct histone acetyl modification defects in a pre-diseased state.
Highly anabolic BRCA1 haploinsufficient cells exhibit increased histone H3 acetylation marks
To characterize the manner of acetyl-CoA-related epigenetic regulation by BRCA1 haploinsufficiency, we used the Luminex® Histone H3 post-translational modification (PTM) assay to simultaneously survey specific acetylation marks (i.e., H3K27, H3K9, and H3K56) as well as the pan-acetylated status of histone H3 in BRCA1185delAG/+ heterozygous and parental BRCA1+/+ cells. We found that BRCA1 haploinsufficient cells exhibited significantly higher H3K27 and H3K56 acetylation levels than the baseline histone acetylation levels found in isogenic BRCA1 parental cells (). Of note, alterations in the histone H3 acetylation marks H3K27ac and H3K56ac, 2 epigenetic markers of active gene transcription, are altered in various cancers. H3K27ac is an active enhancer marker and reflects global cell-type-specific gene expression in various cancer types.Citation47,48 Data show that H3K56ac is increased in multiple types of cancer and is closely related with epigenetic activation of DNA damage response/DNA repair signaling pathways and stem cell pluripotency.Citation49-51 Although the highly abundant H3K9 mark in BRCA1+/+ cells remained unaltered in BRCA1 one-hit cells, a significantly elevated pan-acetylated status of H3 confirmed that an up-regulation of global histone acetylation ostensibly takes place in parallel to the anabolic rewiring occurring in BRCA1 haploinsufficient cells.
Figure 1. Metformin normalizes BRCA1 haploinsufficiency-induced acetylation and phosphorylation of histone H3 marks. BRCA1185delAG/+ heterozygous and parental BRCA1+/+ cells were pretreated with the indicated concentrations of metformin for 48 h. Acid extracts were prepared and lysates were used to evaluate H3 pan-acetyl, H3K27ac, H3K9ac, H3K57ac, H3S10ph, and S3T11ph Ab-conjugated beads in multiplex along with H3 Total beads for normalization to determine relative post-translational modifications (PTM) values using the Active Motif Histone H3 PTM Multiplex Assay. n.s. non-significant differences relative to untreated control cells by Student's t test for paired values; * P < 0.05 relative to control cells by Student's t test for paired values; a: comparing 10 μmol/L metformin (MET)-treated cells to control cells by Student's t test for paired values.
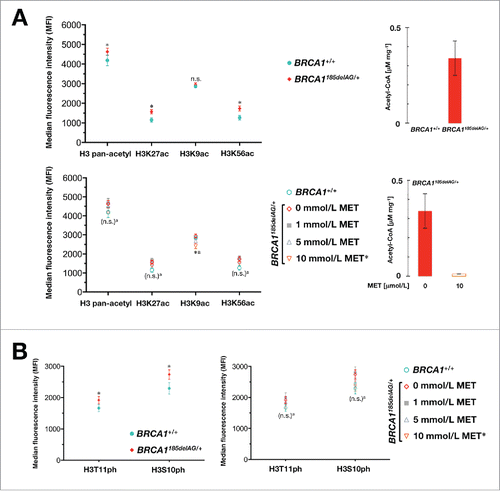
Metformin-driven suppression of acetyl-CoA normalizes histone acetylation marks in BRCA1 haploinsufficient cells
We then evaluated whether the ability of metformin to reverse the anabolic phenotype of BRCA1 one-hit cells by shutting-down mitochondria-driven generation of acetyl-CoA by approximately 90% could decrease the acetylation levels at H3K27 and H3K56. Metformin treatment was sufficient to reduce the abundance of H3K27ac and H3K56ac to levels not significantly different to those of the parental cells (). Moreover, H3K9ac hyperacetylation, which is associated with specific genes in breast cancer,Citation52 was highly sensitive to the inhibitory effects of metformin. Thus, H3K9ac levels were significantly lower in metformin-treated BRCA1 haploinsufficient cells than in BRCA1 wild-type parental cells (). Because phosphorylation of H3 is clearly associated with H3 acetylation, strongly implicating a synergistic coupling of these modifications in transcription activation and the DNA damage response,Citation53-55 we explored whether H3S10 and H3T11 phosphorylation was significantly altered in BRCA1 haploinsufficient cells. BRCA1 one-hit cells likewise exhibited significantly higher levels of H3 phosphorylation on S10 and T11, which were markedly reduced by metformin ().
The route of acetyl-CoA from mitochondria to the nucleus: Where and how metformin regulates histone acetylation
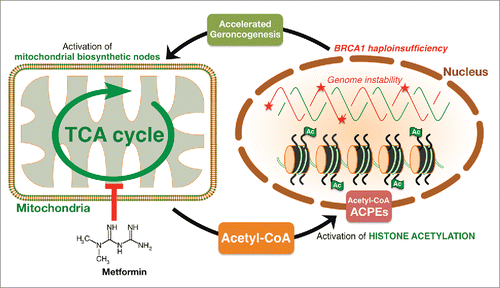
A recent study by the Chandel group has shown how the contribution of mitochondria to control diverse biological outcomes extends beyond their classical bioenergetic function to include proliferation, oxygen sensing, and epigenetics.Citation56 Thus, whereas mitochondrial membrane potential is required for proliferation and HIF-1 activation, the biosynthetic function of the TCA cycle is necessary for histone acetylation. This landmark study demonstrated that mitochondrial respiration regulates specific histone acetylation by allowing the oxidative TCA cycle to generate metabolites.Citation56 We here report that metformin-induced inhibition of mitochondrial respiration regulates specific histone acetylation likely by restricting the biosynthetic capacity of mitochondria to generate the epigenetic metabolite acetyl-CoA ().
It should be acknowledged that the 4 acetyl-CoA-producing enzymes (ACPEs), namely pyruvate dehydrogenase complex (PDC), ATP citrate lyase, acetyl-CoA synthetase short-chain family member 2, and carnitine acetyltransferases, that produce acetyl-CoA in mitochondria or the cytoplasm are all present and functional in the nucleus, producing acetyl-CoA.Citation18 Because the “moonlighting” of ACPEs in the nucleus apparently solves the problem that acetyl-CoA is not permeable through mitochondrial membranes and, owing to its instability, needs to be produced close to where it is needed, it remains to be determined whether the metformin-induced restraining of mitochondrial-dependent biosynthesis of metabolic intermediates imposes either global changes in nuclear acetyl-CoA levels (and therefore regulates global histone acetylation) or small, localized changes in nuclear acetyl-CoA levels (and therefore regulates selective histone acetylation).Citation18 Although the latter scenario appears counterintuitive, it should be noted that pyruvate kinase (PK), especially the isoenzyme PKM2, and mitochondrial PDC are translocated and form a complex in the nucleus with a histone acetyl transferase to locally produce acetyl-CoA and drive specific acetylation of histone marks.Citation57,58 Moreover, nuclear PKM2 operates not only as a biosynthetic enzyme to produce pyruvate to be used by PDC for nuclear generation of acetyl-CoA, but also as a non-canonical kinase that binds and phosphorylates histone H3 at T11 to promote subsequent acetylation of H3 at K9.Citation59 Because metformin can reduce not only pyruvate dehydrogenase activity but also impairs nuclear PKM2 function,Citation60-62 it might be argued that the ability of metformin to target the metabolism-epigenome axis involves the direct effects of ACPEs on histone acetylation/phosphorylation. It is relevant to note that the presence of cytosolic acetate and pyruvate, which can both generate nuclear acetyl-CoACitation57,63 fails to restore histone H3 acetylation marks in cells lacking mitochondrial DNA (rho-zero cells), which suggests that in addition to acetyl-CoA limitation invoked by impaired mitochondrial respiration, there are likely other TCA cycle metabolites that are necessary for histone acetylation.Citation56 Therefore, metformin-limited mitochondrial respiration might regulate specific histone acetylation by impeding the oxidative TCA cycle to generate metabolites.Citation34,35,41
Corollary
By providing the precise metabolic mechanisms for tumor initiation in a tissue-dependent manner, breast epithelial cells from BRCA1 carriers are fertile models of oncogenesis. We recently proposed that the modeling of BRCA1-driven accelerated geroncogenesis might offer an idoneous experimental framework to rapidly and accurately characterize the therapeutic value of metabolo-epigenetic axes that underlie the normal “geroncogenic risk” for those individuals without the mutation.Citation33,41 The identification of the key metabolic messengers directly communicating the metabolic status to the cancer-prone (epi)genome in one-hit BRCA1 breast epithelial cells might uncover unforeseen metabolic strategies to epigenetically target genomic instability and carcinogenesis in more common sporadic forms of age-associated cancers. Using this model of accelerated geroncogenesis, we here confirmed that: 1.) BRCA1 haploinsufficiency-driven metabolic rewiring of the mitochondrial biosynthetic nodes to overproduce epigenetic metabolites such as acetyl-CoA promotes cancer-related acetylation of specific histone H3 marks (e.g., H3K27ac and H3K56ac); 2.) metformin-induced restriction of mitochondrial biosynthetic capacity likewise translates into the epigenetic regulation of histone acetylation. We now provide one of the first examples of how metformin-driven metabolic shifts, such as reduction of the 2-carbon epigenetic substrate acetyl-CoA, is sufficient to correct specific histone H3 acetylation marks in cancer-prone human epithelial cells. The finding that histone acetylation changes following the metabolic stress imposed by metformin supports the notion that directing metabolism to modulate the epigenetic landscape is a promising therapeutic strategy for the management of aging-driven diseases including cancer. Metformin's ability to regulate mitonuclear communicationCitation64 and metabolically modulate the epigenetic landscape should guide the development of new therapeutic strategies involving metabolo-epigenetic aging/cancer prevention and therapy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr. Kenneth McCreath for editorial support.
Funding
Current work in our laboratories are supported by grants from the Plan Nacional de I+D+I, Spain, Instituto de Salud Carlos III (Grant PI15/00285 co-founded by the European Regional Development Fund [FEDER] to JJ, and the Ministerio de Ciencia e Innovación (Grant SAF2012-38914) to JAM. We also acknowledge the support by the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) (Grants 2014 SGR1227 and 2014 SGR229 to JJ and JAM, respectively).
Figure 2. Metformin targets the metabolic control of histone acetylation. The metabolic facet of BRCA1 haploinsufficiency involves alterations in critical metabolic factors for de/methylation, de/acetylation or de/phosphorylation in the nuclear epigenome including acetyl-CoA. Metformin's ability to markedly restrict the production of mitochondrial-dependent biosynthetic intermediates by reducing the anaplerotic flux of glucose, glutamine, and likely branched-chain amino acids into the TCA cycle ultimately impacts on the intracellular concentration of the epigenetic metabolite acetyl-CoA. The change in histone acetylation status occurring in response to metformin might reflect its ability to regulate acetyl-CoA levels. The finding that histone acetylation changes during the metabolic stress imposed by metformin supports the notion that directing metabolism to modulate the epigenetic landscape could be a promising therapeutic strategy for the management of aging-driven diseases including cancer.
Related Research Data
References
- Pavlov NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metabolism 2016; 23:27-47; PMID:26771115; http://dx.doi.org/10.1016/j.cmet.2015.12.006
- Wu LE, Gomes AP, Sinclair DA. Geroncogenesis: Metabolic changes during aging as a driver of tumorigenesis. Cancer Cell 2014; 25:12-19; PMID:24434207; http://dx.doi.org/10.1016/j.ccr.2013.12.005
- Menendez JA, Alarcón T, Joven J. Gerometabolites: The pseudohypoxic aging side of cancer oncometabolites. Cell Cycle 2014; 13:699-709; PMID:24526120; http://dx.doi.org/10.4161/cc.28079
- Menendez JA, Joven J. Energy metabolism and metabolic sensors in stem cells: The metabostem crossroads of aging and cancer. Adv Exp Med Biol 2014; 824:117-140; PMID:25038997; http://dx.doi.org/10.1007/978-3-319-07320-0_10
- Menendez JA. Metabolic control of cancer cell stemness: Lessons from iPS cells. Cell Cycle 2015; 14:3801-11; PMID:25738999; http://dx.doi.org/10.1080/15384101.2015.1022697
- López-Otín C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic control of longevity. Cell 2016; 166:802-821; PMID:Can't; http://dx.doi.org/10.1016/j.cell.2016.07.031
- Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest 2013; 123:3652-8; PMID:23999438; http://dx.doi.org/10.1172/JCI67228
- Nowicki S, Gottlieb E. Oncometabolites: tailoring our genes. FEBS J 2015; 282:2796-805; PMID:25864878; http://dx.doi.org/10.1111/febs.13295
- Morin A, Letouzé E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer 2014; 135:2237-48; PMID:25124653; http://dx.doi.org/10.1002/ijc.29080
- Menendez JA. The metaboloepigenetic dimension of cancer stem cells: Evaluating the market potential for new metabostemness-targeting oncology drugs. Curr Pharm Des 2015; 21:3644-53; PMID:26166605; http://dx.doi.org/10.2174/1381612821666150710150327
- Corrado M, Scorrano L, Campello S. Changing perspective on oncometabolites: from metabolic signature of cancer to tumorigenic and immunosuppressive agents. Oncotarget 2016; 7:46692-706; PMID:27083002
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab 2012; 16:9-17; PMID:22768835; http://dx.doi.org/10.1016/j.cmet.2012.06.001
- Meier JL. Metabolic mechanisms of epigenetic regulation. ACS Chem Biol 2013; 8:2607-21; PMID:24228614; http://dx.doi.org/10.1021/cb400689r
- Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front Oncol 2014; 4:262; PMID:25325014; http://dx.doi.org/10.3389/fonc.2014.00262
- Menendez JA, Corominas-Faja B, Cuyàs E, Alarcón T. Metabostemness: Metaboloepigenetic reprogramming of cancer stem-cell functions. Oncoscience 2014; 1:803-6; PMID:25621295; http://dx.doi.org/10.18632/oncoscience.113
- Menendez JA, Corominas-Faja B, Cuyàs E, García MG, Fernández-Arroyo S, Fernández AF, Joven J, Fraga MF, Alarcón T. Oncometabolic nuclear reprogramming of cancer stemness. Stem Cell Reports 2016; 6:273-83; PMID:26876667; http://dx.doi.org/10.1016/j.stemcr.2015.12.012
- Fernández-Arroyo S, Cuyàs E, Bosch-Barrera J, Alarcón T, Joven J, Menendez JA. Activation of the methylation cycle in cells reprogrammed into a stem cell-like state. Oncoscience 2016; 2:958-67.
- Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic enzymes moonlighting in the nucleus: Metabolic regulation of gene transcription. Trends Biochem Sci 2016; 41:712-30; PMID:27345518; http://dx.doi.org/10.1016/j.tibs.2016.05.013
- Su X, Wellen KE, Rabinowitz JD. Metabolic control of methylation and acetylation. Curr Opin Chem Biol 2016; 30:52-60; PMID:26629854; http://dx.doi.org/10.1016/j.cbpa.2015.10.030
- Dutta A, Abmayr SM, Workman JL. Diverse activities of histone acylations connect metabolism to chromatin function. Mol Cell 2016; 63:547-52; PMID:27540855; http://dx.doi.org/10.1016/j.molcel.2016.06.038
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462:739-44; PMID:19935646; http://dx.doi.org/10.1038/nature08617
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553-67; PMID:21130701; http://dx.doi.org/10.1016/j.ccr.2010.11.015
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483:474-8; PMID:22343901; http://dx.doi.org/10.1038/nature10860
- Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jr, Jahromi MS, Xekouki P, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 2013; 3:648-57; PMID:23550148; http://dx.doi.org/10.1158/2159-8290.CD-13-0092
- Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013; 23:739-52; http://dx.doi.org/10.1016/j.ccr.2013.04.018
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012; 26:1326-38; PMID:22677546; http://dx.doi.org/10.1101/gad.191056.112
- Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal 2014; 26:1598-603; PMID:24704120; http://dx.doi.org/10.1016/j.cellsig.2014.03.030
- Waterfall JJ, Killian JK, Meltzer PS. The role of mutation of metabolism-related genes in genomic hypermethylation. Biochem Biophys Res Commun 2014; 455:16-23; PMID:25111818; http://dx.doi.org/10.1016/j.bbrc.2014.08.003
- Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, Cornelisse CJ, Hogendoorn PC, Breuning MH, Suijker J, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015; 6:38777-88; PMID:26472283; http://dx.doi.org/10.18632/oncotarget.3224
- Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 2015; 3:10; PMID:26401273; http://dx.doi.org/10.1186/s40170-015-0135-3
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab 2015; 22:861-73; PMID:26411344; http://dx.doi.org/10.1016/j.cmet.2015.08.024
- Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci 2016; 1363:91-8; PMID:26647078; http://dx.doi.org/10.1111/nyas.12956
- Menendez JA, Folguera-Blasco N, Cuyàs E, Fernández-Arroyo S, Joven J, Alarcón T. Accelerated geroncogenesis in hereditary breast-ovarian cancer syndrome. Oncotarget 2016; 7:11959-71; PMID:26943589
- Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2014; 2:12; PMID:25184038; http://dx.doi.org/10.1186/2049-3002-2-12
- Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, Jones RG. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol 2015; 13:e1002309; PMID:26625127; http://dx.doi.org/10.1371/journal.pbio.1002309
- Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010; 2:760-74; PMID:21084729
- Menendez JA, Cufí S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressant metformin: less is more. Aging (Albany NY) 2011; 3:348-62; PMID:21483040; http://dx.doi.org/10.18632/aging.100316
- Anisimov VN. Metformin: do we finally have an anti-aging drug? Cell Cycle 2013; 12:3483-9; PMID:24189526; http://dx.doi.org/10.4161/cc.26928
- Anisimov VN. Metformin for cancer and aging prevention: is it a time to make the long story short? Oncotarget 2015; 6:39398-407; PMID:26583576
- Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab 2016; 23:1060-5; PMID:27304507; http://dx.doi.org/10.1016/j.cmet.2016.05.011
- De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L, Temmerman L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci U S A 2014; 111:E2501-9; PMID:24889636; http://dx.doi.org/10.1073/pnas.1321776111
- Cuyàs E, Fernández-Arroyo S, Alarcón T, Lupu R, Joven J, Menendez JA. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: Evidence for metformin-based “starvation” strategies in BRCA1 carriers. Oncotarget 2016; http://dx.doi.org/10.18632/oncotarget.9732
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 2015; 21:805-21; PMID:26039447; http://dx.doi.org/10.1016/j.cmet.2015.05.014
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 2014; 20:306-19; PMID:24998913; http://dx.doi.org/10.1016/j.cmet.2014.06.004
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 2011; 42:426-37; PMID:21596309; http://dx.doi.org/10.1016/j.molcel.2011.05.004
- Galdieri L, Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. J Biol Chem 2012; 287:23865-76; PMID:22580297; http://dx.doi.org/10.1074/jbc.M112.380519
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun 2016; 7:11960; PMID:27357947; http://dx.doi.org/10.1038/ncomms11960
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; http://dx.doi.org/10.1038/nature07829
- Karczmarski J, Rubel T, Paziewska A, Mikula M, Bujko M, Kober P, Dadlez M, Ostrowski J. Histone H3 lysine 27 acetylation is altered in colon cancer. Clin Proteomics 2014; 11:24; PMID:24994966; http://dx.doi.org/10.1186/1559-0275-11-24
- Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem 2010; 285:28553-64; PMID:20587414; http://dx.doi.org/10.1074/jbc.M110.149393
- Tan Y, Xue Y, Song C, Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A 2013; 110:11493-8; PMID:23798425; http://dx.doi.org/10.1073/pnas.1309914110
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009; 459:113-7; PMID:19270680; http://dx.doi.org/10.1038/nature07861
- Sadikovic B, Andrews J, Carter D, Robinson J, Rodenhiser DI. Genome-wide H3K9 histone acetylation profiles are altered in benzopyrene-treated MCF7 breast cancer cells. J Biol Chem 2008; 283:4051-60; PMID:18065415; http://dx.doi.org/10.1074/jbc.M707506200
- Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell 2000; 5:905-15; PMID:10911985; http://dx.doi.org/10.1016/S1097-2765(00)80256-7
- Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 2002; 3:224-9; PMID:11882541; http://dx.doi.org/10.1093/embo-reports/kvf053
- Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012; 7:1098-108; PMID:22948226; http://dx.doi.org/10.4161/epi.21975
- Martínez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell 2016; 61:199-209; http://dx.doi.org/10.1016/j.molcel.2015.12.002
- Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014; 158:84-97; PMID:24995980; http://dx.doi.org/10.1016/j.cell.2014.04.046
- Matsuda S, Adachi J, Ihara M, Tanuma N, Shima H, Kakizuka A, Ikura M, Ikura T, Matsuda T. Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res 2016; 44:636-47; PMID:26405201; http://dx.doi.org/10.1093/nar/gkv967
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012; 150:685-96; PMID:22901803; http://dx.doi.org/10.1016/j.cell.2012.07.018
- Silvestri A, Palumbo F, Rasi I, Posca D, Pavlidou T, Paoluzi S, Castagnoli L, Cesareni G. Metformin induces apoptosis and downregulates pyruvate kinase M2 in breast cancer cells only when grown in nutrient-poor conditions. PLoS One 2015; 10:e0136250; PMID:26291325; http://dx.doi.org/10.1371/journal.pone.0136250
- Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D, et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 2015; 6:24061-74; PMID:26183399; http://dx.doi.org/10.18632/oncotarget.4448
- Salani B, Ravera S, Amaro A, Salis A, Passalacqua M, Millo E, Damonte G, Marini C, Pfeffer U, Sambuceti G, et al. IGF1 regulates PKM2 function through Akt phosphorylation. Cell Cycle 2015; 14:1559-67; PMID:25790097; http://dx.doi.org/10.1080/15384101.2015.1026490
- Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 2015; 33:125-31; PMID:25703630; http://dx.doi.org/10.1016/j.ceb.2015.02.003
- Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 2016; 17:213-26; http://dx.doi.org/10.1038/nrm.2016.23