
ABSTRACT
Cytokines in the developing tumor microenvironment (TME) can drive transformation and subsequent progression toward metastasis. Elevated levels of the Interleukin-6 (IL-6) family cytokine Oncostatin M (OSM) in the breast TME correlate with aggressive, metastatic cancers, increased tumor recurrence, and poor patient prognosis. Paradoxically, OSM engages a tumor-suppressive, Signal Transducer and Activator of Transcription 3 (STAT3)-dependent senescence response in normal and non-transformed human mammary epithelial cells (HMEC). Here, we identify a novel link between OSM-activated STAT3 signaling and the Transforming Growth Factor-β (TGF-β) signaling pathway that engages senescence in HMEC. Inhibition of functional TGF-β/SMAD signaling by expressing a dominant-negative TGF-β receptor, treating with a TGF-β receptor inhibitor, or suppressing SMAD3 expression using a SMAD3-shRNA prevented OSM-induced senescence. OSM promoted a protein complex involving activated-STAT3 and SMAD3, induced the nuclear localization of SMAD3, and enhanced SMAD3-mediated transcription responsible for senescence. In contrast, expression of MYC (c-MYC) from a constitutive promoter abrogated senescence and strikingly, cooperated with OSM to promote a transformed phenotype, epithelial-mesenchymal transition (EMT), and invasiveness. Our findings suggest that a novel STAT3/SMAD3-signaling axis is required for OSM-mediated senescence that is coopted during the transformation process to confer aggressive cancer cell properties. Understanding how developing cancer cells bypass OSM/STAT3/SMAD3-mediated senescence may help identify novel targets for future “pro-senescence” therapies aiming to reengage this hidden tumor-suppressive response.
Introduction
Cancer development is driven by genetic and epigenetic changes within a cell, as well as extrinsic factors in the microenvironment. Cytokines within the tumor microenvironment (TME), often generated due to chronic inflammation, can drive tumor progression and metastasis.Citation1-3 The Interleukin-6 (IL-6) family of pro-inflammatory cytokines, which includes Oncostatin M (OSM), regulate various biological processes, and deregulated IL-6 family signaling is often characteristic of more aggressive cancers.Citation2,4-11 OSM-mediated signal transduction occurs through hetero-dimerization of glycoprotein 130 (gp130) and OSM receptor subunit β (OSMRβ).Citation12 The OSMR complex activates Janus Kinases 1 and 2 (JAK1 and JAK2), which in turn activate Signal Transducer and Activator of Transcription 3 (STAT3), mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K)-AKT-mediated signaling cascades.Citation13-18 Elevated levels of OSM in the TME are associated with highly aggressive metastatic cancers, increased risk of tumor recurrence, and a poor prognosis.Citation19-24 In breast cancer, OSM is concentrated at the invasive edges of highly metastatic tumors where cells often display mesenchymal cell characteristics and express the cancer stem cell marker CD44.Citation22,25
Whereas OSM drives proliferation, epithelial-mesenchymal transition (EMT), invasion, and metastasis in breast cancer cells and transformed human mammary epithelial cells (HMEC), it engages senescence in normal and non-transformed HMEC.Citation13,19-21,26-34 Senescence is an irreversible, proliferation-inhibiting response that occurs in 2 steps: 1) reversible cell cycle arrest followed by 2) futile growth-induced gerogenic conversion, or geroconversion.Citation35-37 Moreover, senescence is a major tumor-suppressive mechanism that acts as an early barrier to thwart cancer development. The presence of senescent cells in benign lesions, but not advanced malignant tumors, suggests that senescence barriers must be dismantled during oncogenic progression.Citation38 In the case of OSM-induced senescence, persistent STAT3 activation represses MYC (c-MYC) expression and engages a p16- and p53-independent cell cycle arrest that is accompanied by many senescence-associated characteristics, including an enlarged flattened morphology and β-galactosidase (β-Gal) activity.Citation19,39,40 While OSM-induced MYC repression is associated with senescence, ectopic expression of MYC from a constitutive promoter prevents senescence and alters the response to OSM from tumor-suppressive to tumor-promoting.Citation19 Understanding how developing tumor cells adapt to senescence-inducing cytokine signals and circumvent the normal, growth-suppressive response is critical for understanding tumorigenesis.
Using isogenic HMEC, we now reveal that OSM/STAT3-induced senescence requires Transforming Growth Factor-β (TGF-β)/SMAD3 signaling. OSM/STAT3 signaling induces a STAT3/SMAD3 interaction, increases SMAD3 nuclear localization, enhances SMAD3-mediated transcription, and ultimately suppresses proliferation. Dysregulated MYC expression dismantles STAT3/SMAD3-induced senescence and allows OSM-mediated STAT3/SMAD3 signaling to then drive anchorage-independent growth (AIG), EMT, and invasion. Our studies have direct relevance for understanding how senescence pathways are suppressed during the transformation process, and provide a foundation for novel “pro-senescence” therapies aimed at reengaging hidden tumor-suppressive responses.
Results
OSM/STAT3-induced senescence requires TGF-β signaling
Previous studies using normal human mammary epithelial cells (HMEC) have demonstrated that oncogenic RAS (RAS-G12V) and RAF1 induce oncogene-induced senescence (OIS) in a p16- and p53-independent manner.Citation41,42 Likewise, persistent Oncostatin M (OSM)-mediated activation of STAT3 also induces a p16/p53-independent senescence.Citation19 Given that RAS-induced OIS requires functional Transforming Growth Factor-β (TGF-β) signaling, we hypothesized that persistent OSM/STAT3-induced senescence would also utilize the TGF-β signaling pathway.Citation41 To test this hypothesis, post-selection shp53-HMEC (lacking p16 expression due to endogenous p16 promoter methylation and p53 due to shRNA-mediated ablation) were infected with retroviruses encoding either a dominant-negative STAT3 (shp53/DN-STAT3-HMEC) or a dominant-negative TGF-β type II receptor (shp53/DN-TGFβR2-HMEC) to inhibit TGF-β signaling. An empty vector (shp53/vector-HMEC) was used as a control. Each of the shp53-HMEC derivatives were treated with recombinant OSM or recombinant TGF-β1 for 7 days, and cell number was quantified. Whereas treatment with OSM or TGF-β1 reduced the growth of shp53/vector-HMEC by approximately 75% in comparison to untreated cells, DN-TGFβR2 expression largely prevented both OSM- and TGF-β1-mediated growth reduction (). Likewise, as previously reported, DN-STAT3 expression prevented OSM-, but not TGF-β1-induced growth suppression.Citation19 Western analysis of whole cell lysates using an antibody directed at the OSMR-mediated phosphorylation site of STAT3 (Tyrosine 705) confirmed that STAT3 phosphorylation (P-STAT3) was increased in OSM-treated control shp53/vector-HMEC and shp53/DN-TGFβR2-HMEC, but suppressed in OSM-treated shp53/DN-STAT3-HMEC (). Similarly, Western analysis using an antibody directed at the TGFβR-mediated phosphorylation site of SMAD2 (Serine 465/467) confirmed that SMAD2 phosphorylation (P-SMAD2) was increased in shp53/vector-HMEC and shp53/DN-STAT3-HMEC, but suppressed in shp53/DN-TGFβR2-HMEC, following TGF-β1-treatment ().
Figure 1. OSM/STAT3-induced senescence requires TGF-β signaling. (A) Relative growth assays (top) and western analysis of whole cell lysates (bottom) of shp53/vector-, DN-STAT3-, or DN-TGFβR2-HMEC plated in the presence (+) and absence (-) of recombinant OSM [10 ng/mL] or TGF-β1 [5 ng/mL] for 7 d. (B) Relative growth assays (top) and western analysis (bottom) of shp53-HMEC treated with recombinant OSM alone or in combination with one of 2 different small molecule TGFβR1 inhibitors, SB525334 [1 μM] (left) and SB431542 [5 μM] (right) for 7 d.
![Figure 1. OSM/STAT3-induced senescence requires TGF-β signaling. (A) Relative growth assays (top) and western analysis of whole cell lysates (bottom) of shp53/vector-, DN-STAT3-, or DN-TGFβR2-HMEC plated in the presence (+) and absence (-) of recombinant OSM [10 ng/mL] or TGF-β1 [5 ng/mL] for 7 d. (B) Relative growth assays (top) and western analysis (bottom) of shp53-HMEC treated with recombinant OSM alone or in combination with one of 2 different small molecule TGFβR1 inhibitors, SB525334 [1 μM] (left) and SB431542 [5 μM] (right) for 7 d.](/cms/asset/68af2cf1-2706-49d0-93e7-c3fc489a845c/kccy_a_1259037_f0001_b.gif)
TGF-β ligand binding stimulates the assembly of TGFβR-complexes composed of TGF-β type I (TGFβR1) and TGF-β type II receptors (TGFβR2). The TGFβR-complex becomes activated after TGFβR2 phosphorylates and triggers the protein kinase activity of TGFβR1, which is then responsible for propagating the TGF-β signal. Thus, inhibiting the activity of either TGFβR1 or TGFβR2 is sufficient to inhibit downstream TGFβR signaling. As an alternative approach to determine whether functional TGFβR signaling is required for OSM/STAT3-induced senescence, shp53-HMEC were treated for 7 d with recombinant OSM alone or in combination with one of 2 different small molecule TGFβR1 inhibitors, SB525334 and SB431542. As observed following the expression of DN-TGFβR2, OSM-induced growth reduction was abrogated by co-treatment with either SB525334 or SB431542 (). Neither SB525334 nor SB431542 impacted OSM-induced P-STAT3, but did reduce basal P-SMAD2 levels (). Taken together, our results indicate that OSM/STAT3-induced senescence requires functional TGFβR signaling and that an effector downstream of JAK-mediated STAT3 phosphorylation is suppressed by TGFβR inhibition.
OSM/STAT3-induced senescence requires SMAD3/SMAD4
Canonical TGF-β-induced senescence in normal HMEC is engaged by receptor-regulated SMADs (R-SMADs, specifically SMAD2 and SMAD3) that are activated by TGFβR1-mediated phosphorylation.Citation43 Once phosphorylated, SMAD2 and SMAD3 bind to the co-SMAD, SMAD4, in the cytoplasm and then translocate into the nucleus. The TGFβR1-mediated activation and nuclear localization of SMAD2 and SMAD3 is prevented by an inhibitory SMAD (I-SMAD), SMAD7 (). SMAD2/SMAD4 and SMAD3/SMAD4 complexes transcriptionally regulate a number of target genes containing SMAD-binding elements (SBE), including the cyclin-dependent kinase inhibitors, p15 and p21, which prevent CyclinD1/CDK activity and engage senescence ().Citation43 Since inhibiting functional TGFβR signaling with DN-TGFβR2 and small molecule TGFβR1 inhibitors was sufficient to abrogate OSM/STAT3-induced senescence, we sought to determine whether SMAD activity downstream of TGF-βR was required for OSM/STAT3-induced senescence. To test this, shp53-HMEC were infected with retroviruses encoding SMAD7 (shp53/SMAD7-HMEC) or a constitutively active CyclinD1/CDK2 fusion protein (shp53/CA-D1/CDK2-HMEC). Again, OSM induced a strong growth suppression in shp53/vector-HMEC, which was prevented in shp53/DN-STAT3-HMEC and shp53/DN-TGFβR2-HMEC. Importantly, OSM-mediated growth suppression was abrogated in both shp53/SMAD7-HMEC and shp53/CA-D1/CDK2-HMEC (, top). Again, Western analysis demonstrated that the levels of P-STAT3 in OSM-treated shp53/SMAD7-HMEC and shp53/CA-D1/CDK2-HMEC were comparable to those in shp53/vector-HMEC (, bottom).
Figure 2. OSM/STAT3-induced senescence requires SMAD3/SMAD4. (A) Schematic of canonical TGF-β/SMAD signaling-induced senescence in normal HMEC. (B) Relative growth assays (top) and Western analysis (bottom) of shp53-HMEC expressing either empty vector, DN-STAT3, DN-TGFβR2, SMAD7, or a constitutively-active Cyclin D1/CDK2 fusion protein (CA-D1/CDK2) plated in the presence (+) and absence (-) of recombinant OSM for 7 d. (C) Relative growth assays (left) and western analysis (right) of shp53-HMEC stably expressing shRNAs targeting either green fluorescent protein (shGFP), SMAD2 (shSMAD2), SMAD3 (shSMAD3), or SMAD4 (shSMAD4) plated in the presence (+) and absence (-) of recombinant OSM for 7 d.
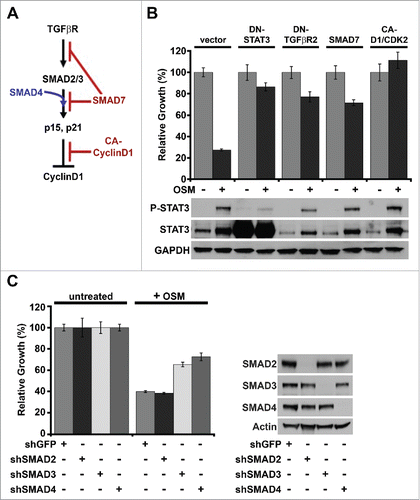
Both SMAD7 and the CA-CyclinD1/CDK2 fusion protein act as global inhibitors to the cytostatic activity of R-SMADs downstream of the TGF-β receptor. Thus, we next sought to delineate which downstream SMADs were required for OSM/STAT3-induced senescence by infecting shp53-HMEC with retroviruses encoding shRNAs targeting either SMAD2, SMAD3, SMAD4, or green fluorescent protein (shp53/shGFP-HMEC) as a control. Interestingly, the OSM-induced growth reduction was inhibited in both shp53/shSMAD3-HMEC and shp53/shSMAD4-HMEC, but not shp53/shSMAD2-HMEC (, left). Western analysis confirmed the specific shRNA-mediated suppression of SMAD expression in each shp53/shSMAD-HMEC-derivative (, right). Taken together, these data indicate that OSM/STAT3-induced senescence requires the canonical TGF-β-regulated changes in gene transcription that are mediated specifically by SMAD3/SMAD4 complexes downstream of TGF-β receptor.
OSM/STAT3 signaling promotes SMAD3 nuclear localization without increased phosphorylation
Given the importance of SMAD3/SMAD4 complexes in OSM/STAT3-induced senescence, we hypothesized that persistent OSM/STAT3 signaling promotes increased TGFβR1-mediated phosphorylation, and thus canonical activation of SMAD3. To test this hypothesis, shp53-HMEC were treated for 4 and 8 d with either OSM or TGF-β1 as a control. Western analysis of whole cell lysates was used to assess P-STAT3 and TGFβR1-mediated phosphorylation of SMAD2, and SMAD3 (Serine 423/425). As expected, P-STAT3 was increased following exposure to OSM and P-SMAD2 and P-SMAD3 were increased following TGF-β1 exposure. However, despite the need for functional TGFβR signaling during OSM-mediated senescence ( and ), persistent OSM/STAT3 signaling did not significantly alter P-SMAD2 or P-SMAD3 levels ().
Figure 3. OSM/STAT3 signaling promotes SMAD3 nuclear localization without increased phosphorylation. (A) Western analysis of whole cell lysates harvested from shp53-HMEC plated in the presence (+) and absence (-) of either OSM or TGF-β1 for 4 and 8 d. (B) Western analysis of the nuclear-soluble protein fractions from subcellular fractionations of untreated shp53-HMEC and shp53-HMEC treated with either OSM or TGF-β1 for 4 d. (C) Western analysis of SMAD3-specific immunoprecipitations using whole cell lysates from both untreated and OSM-treated HEK 293T cells transfected with an expression vector encoding SMAD3 in combination with either an expression vector encoding wild-type STAT3 (WT-STAT3) or encoding dominant-negative STAT3 (DN-STAT3), and then plated in the presence (+) and absence (-) of recombinant OSM [10 ng/mL] for 2 d.
![Figure 3. OSM/STAT3 signaling promotes SMAD3 nuclear localization without increased phosphorylation. (A) Western analysis of whole cell lysates harvested from shp53-HMEC plated in the presence (+) and absence (-) of either OSM or TGF-β1 for 4 and 8 d. (B) Western analysis of the nuclear-soluble protein fractions from subcellular fractionations of untreated shp53-HMEC and shp53-HMEC treated with either OSM or TGF-β1 for 4 d. (C) Western analysis of SMAD3-specific immunoprecipitations using whole cell lysates from both untreated and OSM-treated HEK 293T cells transfected with an expression vector encoding SMAD3 in combination with either an expression vector encoding wild-type STAT3 (WT-STAT3) or encoding dominant-negative STAT3 (DN-STAT3), and then plated in the presence (+) and absence (-) of recombinant OSM [10 ng/mL] for 2 d.](/cms/asset/3e9026fb-8960-45e1-9078-12236e717003/kccy_a_1259037_f0003_b.gif)
Since SMAD2 and SMAD3 function as transcription factors, we next assessed whether persistent OSM/STAT3 signaling induced SMAD nuclear translocation by using sub-cellular fractionation. Western analysis of protein extracts from nuclear-soluble fractions assessed the nuclear localization of STAT3, SMAD2, and SMAD3, and also protein phosphorylation of STAT3 (P-STAT3) and SMAD3 (P-SMAD3). Importantly, the nuclear localization of total SMAD3 was increased following OSM-treatment and paralleled the increased nuclear localization of total SMAD3 induced by TGF-β1-treatment (). In contrast, the nuclear localization of SMAD2 was not altered following treatment with OSM (), consistent with our previous results demonstrating that the loss of SMAD2 does not block OSM-induced growth reduction (). As expected, the nuclear localization of P-STAT3 and total STAT3 was increased by OSM but not by TGF-β1 (). Our results demonstrate that both STAT3 and SMAD3 are required for OSM-induced senescence and that the nuclear localization of both STAT3 and SMAD3 is induced by OSM.
Both STAT3 and SMAD3 function as transcription factors in the nucleus and are known to complex with additional transcription factors to control gene expression. To assess whether STAT3 and SMAD3 form a complex, cells were co-transfected with STAT3 and SMAD3 expression vectors, and treated with OSM prior to co-immunoprecipitation/Western analysis. WT-STAT3 co-precipitated with SMAD3 in both untreated and OSM-treated cell lysates (). Moreover, the level of STAT3 co-precipitated with SMAD3 was higher in OSM-treated cells compare with untreated cells (). Co-precipitation of DN-STAT3 with SMAD3 was greatly reduced when compare with WT-STAT3, suggesting that STAT3-SMAD3 complexes are facilitated by OSM-mediated STAT3 phosphorylation and dimerization. Taken together, our results suggest that persistent OSM/STAT3 signaling induces a STAT3-SMAD3 interaction that can promote the nuclear localization of SMAD3, a key determinant of OSM-mediated senescence.
Persistent OSM/STAT3 signaling promotes SMAD target-gene transcription
Since SMAD3 functions as a transcription factor within the nucleus, we hypothesized that STAT3-mediated SMAD3 nuclear localization would promote SMAD3-mediated gene expression. To test this hypothesis, mRNA was harvested from untreated, OSM, or TGF-β1 treated shp53-HMEC and the expression of TGF-β pathway genes was assessed using a TGF-β signaling targets qRT-PCR profiler array. Genes exhibiting a similar change in expression at least 2-fold in response to both OSM and TGF-β1 were selected and plotted. Importantly, the expression of 36 out of 84 (43%) genes was changed more than 2-fold following TGF-β1 treatment. The expression of 31 of these 36 genes (86% overlap) were similarly changed more than 2-fold following OSM treatment (). To confirm the results from the TGF-β qRT-PCR profiler array, the expression of 2 genes upregulated by OSM (SNAI1 and TNFSF10) and 2 genes repressed by OSM (MYC and ATF3) was confirmed by qRT-PCR. The expression of both SNAI1 and TNFSF10 was upregulated ∼8-fold in response to OSM treatment, and the upregulated expression was abrogated by co-treatment with SB431542 (). Likewise, MYC was repressed 3-fold following OSM-treatment, which was again abrogated by co-treatment with SB431542 (). Similarly, ATF3 was repressed 10-fold, consistent with the repression observed in the qRT-PCR profiler array, however, the OSM-induced repression of ATF3 was not affected by SB431542 treatment ().
Figure 4. Persistent OSM/STAT3 signaling promotes SMAD target-gene transcription. (A) mRNA harvested from shp53-HMEC plated in the presence (+) and absence (-) of either recombinant OSM [10 ng/mL] or recombinant TGF-β1 [5 ng/mL] for 7 d was subjected to a targeted TGF-β Signaling-Targets qRT-PCR profiler array. Genes exhibiting a similar change in expression at least 2-fold following treatment with both OSM and TGF-β1 were selected and displayed to compare gene expression between untreated shp53-HMEC and shp53-HMEC treated with either OSM or TGF-β1. (B) Single-gene qRT-PCR using mRNA harvested from shp53-HMEC left untreated or treated with either OSM alone or in combination with the small molecule TGFβR1 inhibitor, SB431542, and using primers targeting SNAI1, TNFSF10, MYC, and ATF3. (C) Single-gene qRT-PCR using primers targeting SNAI1 and mRNA from shp53-HMEC expressing shRNAs to GFP (shGFP), SMAD2 (shSMAD2), SMAD3 (shSMAD3), or SMAD4 (shSMAD4) grown in the presence (+) or absence (-) of recombinant OSM for 7 d.
![Figure 4. Persistent OSM/STAT3 signaling promotes SMAD target-gene transcription. (A) mRNA harvested from shp53-HMEC plated in the presence (+) and absence (-) of either recombinant OSM [10 ng/mL] or recombinant TGF-β1 [5 ng/mL] for 7 d was subjected to a targeted TGF-β Signaling-Targets qRT-PCR profiler array. Genes exhibiting a similar change in expression at least 2-fold following treatment with both OSM and TGF-β1 were selected and displayed to compare gene expression between untreated shp53-HMEC and shp53-HMEC treated with either OSM or TGF-β1. (B) Single-gene qRT-PCR using mRNA harvested from shp53-HMEC left untreated or treated with either OSM alone or in combination with the small molecule TGFβR1 inhibitor, SB431542, and using primers targeting SNAI1, TNFSF10, MYC, and ATF3. (C) Single-gene qRT-PCR using primers targeting SNAI1 and mRNA from shp53-HMEC expressing shRNAs to GFP (shGFP), SMAD2 (shSMAD2), SMAD3 (shSMAD3), or SMAD4 (shSMAD4) grown in the presence (+) or absence (-) of recombinant OSM for 7 d.](/cms/asset/bf011f87-dedb-4fe3-9758-6342a6020fe8/kccy_a_1259037_f0004_b.gif)
Since OSM-mediated growth suppression was dependent on SMAD3 and SMAD4 (), we hypothesized that SNAI1 induction following OSM-treatment () would also require SMAD3 and SMAD4. To test this hypothesis, SNAI1 expression was assessed in shSMAD2, shSMAD3, and shSMAD4-expressing shp53/HMEC following treatment with OSM. Indeed, the upregulated SNAI1 expression induced by OSM was strongly inhibited following ablation of either SMAD3 or SMAD4, but not SMAD2 (). Our findings demonstrate that OSM induces a gene expression signature that is markedly similar to TGF-β, and that basal TGFβR signaling is required for OSM-mediated SNAI1 and TNFSF10 induction and MYC repression. Moreover, our results indicate that the OSM-induced changes in SMAD-target gene expression require transcriptional activities that are mediated specifically by SMAD3/SMAD4 complexes.
Constitutive MYC expression dismantles STAT3/SMAD3-induced senescence and cooperates with OSM to drive EMT and invasiveness
MYC is one of the most frequently dysregulated oncogenes and is commonly overexpressed in many types of human cancer, including breast cancer.Citation44-55 Both OSM/STAT3- and TGF-β-induced senescence require the repression of MYC.Citation40,56-59 Our lab has previously demonstrated that the expression of MYC from a constitutive promoter prevents OSM- or RAS-induced senescence and alters the response of HMEC to persistent oncogenic stimuli, from growth suppressive to growth promoting.Citation19,41 Moreover, after dismantling the tumor suppressive senescence barrier, MYC expression cooperates with persistent OSM or RAS signaling to drive transformation.Citation19,41 Thus, similar to the reported “TGF-β paradox,” we hypothesized that once OSM/STAT3-induced senescence was dismantled by constitutive MYC expression, persistent OSM-induced STAT3/SMAD3 signaling would promote phenotypic traits associated with malignant progression (anchorage-independent growth, epithelial-mesenchymal transition (EMT), and invasive properties). To test this hypothesis, MYC expressing HMEC (shp53/MYC-HMEC) were treated with either recombinant OSM or TGF-β1 and plated into soft agar to assess anchorage independent growth (AIG), a characteristic associated with cellular transformation. As reported previously, shp53/MYC-HMEC are incapable of AIG, however, treatment with either OSM or TGF-β1 promoted robust AIG (). To determine whether OSM signaling drives EMT shp53/MYC-HMEC were treated with OSM (or TGF-β1 as a control), protein and mRNA was harvested, and markers of EMT were assessed by qRT-PCR (CDH1 and SNAI1 mRNA) or Western analysis (E-cadherin and Vimentin). E-cadherin (encoded by CDH1), a gene expressed in epithelial cells and downregulated during EMT, was repressed by OSM and TGF-β1 in both shp53-HMEC and shp53/MYC-HMEC (). In contrast, Snail, a transcription factor that promotes and is upregulated during EMT, and Vimentin, an intermediate filament protein that is expressed in mesenchymal cells, were induced by OSM and TGF-β1 in both shp53-HMEC and shp53/MYC-HMEC (, respectively). The downregulation of CDH1 concomitant with upregulation of SNAI1 and Vimentin indicates that the OSM-induced STAT3/SMAD3 signaling observed during senescence can be co-opted to induce EMT once senescence barriers have been abrogated. Finally, since persistent OSM signaling induced EMT, we hypothesized that persistent OSM exposure would also promote an invasive phenotype. To test this hypothesis, shp53/MYC-HMEC were plated into organotypic culture in the presence or absence of OSM and grown for 2 weeks. The development of an invasive phenotype was visually characterized by confocal and bright field microscopy. Untreated shp53/MYC-HMEC grown in organotypic cultures formed well-organized spheroids that highly express the epithelial marker, E-cadherin and lack the mesenchymal marker Vimentin (). Persistent exposure to OSM induced a highly invasive phenotype characterized by cellular outgrowths that detached and migrated away from the periphery in ∼40% of the spheroids (). Moreover, the OSM-induced invasive outgrowths occurred concomitantly with the downregulation of E-cadherin and upregulation of Vimentin (). Importantly, the untreated shp53/MYC-HMEC only formed well-organized spheroids lacking invasive outgrowths (). Thus, we conclude that cooperation between persistent OSM signaling and MYC promotes EMT resulting in an invasive phenotype.
Figure 5. Constitutive MYC expression dismantles STAT3/SMAD3-induced senescence and cooperates with OSM to drive EMT. (A) Soft agar culture of shp53/MYC-HMEC plated in the presence or absence of recombinant OSM and recombinant TGF-β1 for 2 weeks. AIG was quantified by brightfield microscopy using Metamorph software. (B) Single-gene qRT-PCR analysis using primers targeting E-cadherin and Snail (encoded by CDH1 and SNAI1, respectively) and mRNA from shp53-HMEC and shp53/MYC-HMEC plated in the presence (+) or absence (-) of recombinant OSM and TGF-β1 for 7 d. (C) Western analysis of whole cell lysates from shp53/MYC-HMEC left untreated or treated with recombinant OSM for 7 d.
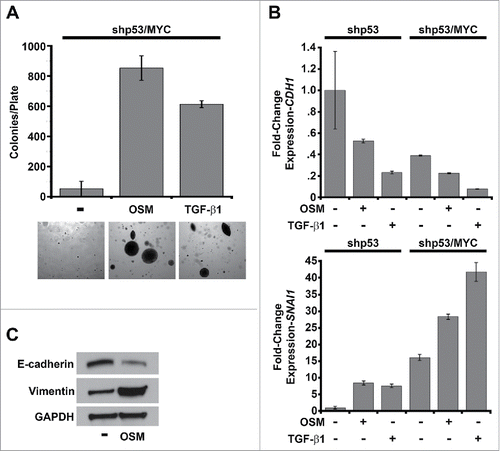
Figure 6. Constitutive MYC expression dismantles STAT3/SMAD3-induced senescence and cooperates with OSM to drive invasiveness. Shp53/MYC-HMEC were plated in 3D laminin-rich basement membrane (matrigel) culture in the presence and absence of recombinant OSM for 10 d. (A) Immunofluorescence using confocal microscopy at 100X magnification with Alexafluor-conjugated antibodies directed against E-cadherin (red) or Vimentin (green) and DAPI stain (blue, nuclear stain). (B) Invasive acini were photographed and quantified by brightfield microscopy at 5X magnification. (C) Following OSM-treatment, mRNA was harvested from shp53/MYC-HMEC after extraction from 3D matrigel culture and then subjected to a targeted Extracellular Matrix (ECM) and Adhesion Molecules qRT-PCR profiler array to compare gene expression differences between untreated and OSM-treated cells.
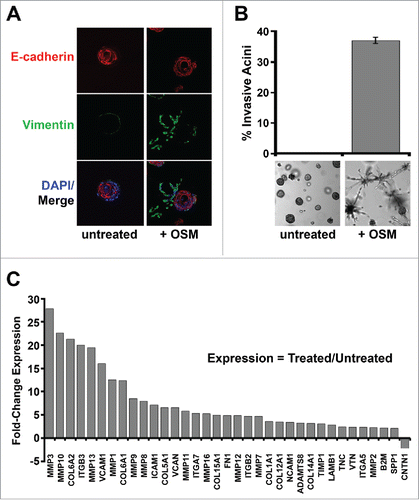
The EMT program and invasion into adjacent tissues by transformed epithelial cells involves dynamic transcriptional change of genes encoding proteins that regulate cell-cell and cell-extracellular matrix (ECM) interactions and the remodeling of surrounding ECM. We next assessed the effect of OSM on the transcription of genes encoding ECM and adhesion molecules. RNA was harvested from untreated and OSM-treated shp53/MYC-HMEC extracted from organotypic culture after 2 weeks and subjected to qRT-PCR using a targeted Extracellular Matrix and Adhesion Molecules profiler array. Of the 84 genes on the array, 34 (40%) were upregulated at least 2-fold in shp53/MYC-HMEC exposed to OSM (). In addition to the upregulation of ECM proteins and adhesion molecules, numerous ECM remodeling-proteases, such as the matrix metalloproteases (MMP1, MMP2, MMP3, MMP7, MMP8, MMP9, MMP10, MMP11, MMP12, MMP13, MMP16), were also upregulated following exposure to OSM (). Taken together, these results indicate that, after OSM/STAT3-induced senescence is dismantled by constitutive MYC expression, persistent OSM signaling promotes AIG, EMT, and invasion to drive cancer development and progression toward metastasis ().
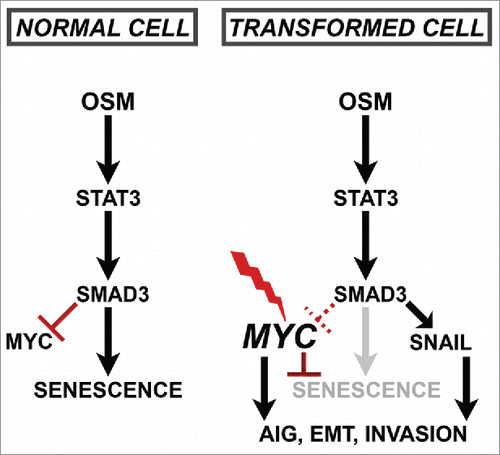
Figure 7. Schematic of Proposed Paradoxical OSM/STAT3/SMAD3-induced Mechanisms.
Discussion
Senescence is a key tumor-suppressive mechanism that acts as an early barrier to oncogenic transformation. The presence of senescent cells in benign, but not advanced malignant tumors, suggests that the pathways leading to senescence are dismantled during full oncogenic progression.Citation60-68 Our results provide important insight into how persistent OSM signaling utilizes TGF-β effectors to induce senescence and prevent transformation in non-transformed epithelial cells. First noted following expression of mutant oncogenes such as RAS and RAF1, oncogene-induced senescence (OIS) involves a cell intrinsic response to replicative stress induced by inappropriate, forced proliferation.Citation41,42 Notably, oncogenes that engage OIS simultaneously induce cell cycle inhibitors, such as p16 and p21, while activating strong mitogenic signals, such as the MAPK and PI3K-AKT pathways; and as a result, initiate 2 processes involved in the senescence program: cell cycle arrest and geroconversion.Citation35,37,69 It was postulated that OIS prevents incipient cancer cells harboring oncogenic mutations from becoming fully tumorigenic. Our study asked the intriguing question: can OIS be engaged by persistent hyperactivation of an oncogene (STAT3), elicited by inappropriate, paracrine activation by extrinsic factors rather than intrinsic genetic changes? The foundation for this question is rooted in the observation that leukocyte infiltrates surround developing neoplasms; an observation that is viewed by many as an attempt by the immune system to suppress tumorigenesis.Citation70 While the OSM protein is not detectable in healthy human tissues, it is frequently expressed during inflammatory conditions (i.e. cancer, arthritis, inflammatory heart disease) due to its secretion from neutrophils, dendritic cells, monocytes and macrophages.Citation22,71-75 Thus, we propose that OSM would be prevalent in the leukocytic infiltrates surrounding developing neoplasms in vivo, providing an important relevance to our studies.
Similar to our findings with persistent OSM/STAT3 signaling presented here, we previously reported that OIS engaged by oncogenic RAS signaling utilizes the TGF-β pathway.Citation41 Importantly, our previous studies demonstrate that OSM is considerably more potent at inducing senescence when compare with IL-6, although the reasons behind this difference is not clear.Citation19 OSM activates a gp130/OSMRβ receptor (OSMR) protein complex that is distinct from other IL-6 family co-receptors, which may account for the more potent OSM-induced senescence response.Citation26,76,77 OSMR complexes consisting of gp130/OSMRβ heterodimers can activate STAT3, RAS-MAPK, and PI3K-AKT signaling, in addition to pathways not activated by other IL-6 family receptor complexes that signal via gp130/gp130 homodimers (STAT5, STAT6, AKT, and PKC-δ).Citation19,78 Moreover, OSMR recruits Shc, which can help to hyper-activate MAPK signaling beyond what is achieved by other gp130 signaling complexes.Citation76
In order to bypass the senescence program, developing cancer cells must dismantle cell cycle control by either inhibiting p16, pRb, and p53 or activating MYC (c-MYC).Citation35,37 Central to the senescence program that is engaged following either oncogenic RAS expression or persistent OSM/STAT3 activation is the repression of endogenous MYC (c-myc) expression.Citation19 Therefore, cell cycle arrest and geroconversion to irreversible senescence that is mediated by either oncogenic RAS or persistent OSM/STAT3 signaling can be abrogated simply by preventing MYC repression.Citation19,41 This can be achieved experimentally by expressing MYC from an orthologous promoter incapable of being repressed. Importantly, MYC expression is commonly dysregulated in breast cancers and correlates with more aggressive tumors and poor patient outcomes.Citation47,48,51,53 Interestingly, whereas mutations in TGFβR or SMAD proteins are common in other cancers, they are not common in breast cancer.Citation79 We propose that dysregulated MYC expression during cancer development serves to (1) prevent senescence; and (2) allows developing tumor cells to retain the functional TGF-β/SMAD signaling components responsible for the more invasive, mesenchymal phenotypes observed here (, right). Indeed, Snail is a key EMT-inducing transcription factor identified here as one of the many OSM/STAT3/SMAD3-regulated genes, in addition to a number of MMPs and extracellular matrix proteins (, right).
In addition to the frequent dysregulation of MYC, OSM is itself significantly elevated in breast cancer, as well as many other cancers.Citation14,19,29,31,34,78,80-86 We propose that the elevated OSM expression levels observed in cancerous tissues is an extension of the inflammatory environment initially created during the early stages of transformation as a means to suppress tumorigenicity. There is mounting evidence that OSM in the TME contributes to tumor progression.Citation1,19-24,28,33,74,87-93 For example, in addition to the increased OSM levels in the breast TME, breast cancer cells also display elevated OSMR expression levels, which is associated with adverse clinical outcomes.Citation32,71,83,88,92,94 Notably, the more aggressive basal-like or triple-negative breast cancer subtype expresses markedly higher levels of OSMR when compare with the less aggressive luminal subtype.Citation71 Moreover, elevated OSMR expression correlates with increased expression of the cancer stem cell (CSC) marker CD44.Citation71 Coupled with these findings is the observation that OSM is secreted by macrophages localized at the advancing, infiltrative margins of carcinomas, where invasive tumor cells reside and intravasate during metastasis.Citation22 The unique signaling that emanates from OSMRβ/gp130 receptor complexes discussed above may explain why elevated OSM in the TME correlates with more invasive, metastatic tumors, as CD44-expressing cancer cells, recently classified as metastasis-initiating cells, are also found primarily at the invasive edge of tumors.Citation22,71 In addition, DNA damaging chemotherapy induces additional OSM secretion from macrophages, potentially increasing mesenchymal and CSC properties following treatment with genotoxic therapies.Citation90,95,96 When found together in tumors, dysregulated MYC and increased OSM create the potential for invasive, metastatic, and potentially therapy-resistant cancer cells. Our findings suggest that targeting MYC would re-engage the STAT3/SMAD3-mediated senescence, making it an ideal therapeutic target (). However, due to its importance in normal cell physiology, MYC is unfortunately considered an undruggable therapeutic target. Likewise, while blocking STAT3 activation using JAK or STAT3 inhibitors in combination with chemotherapy may suppress some of the unintended consequences of chemotherapies (such as creating resistant cells with greater invasive potential), it may also release dormant pre-malignant cells from their tumor suppressive constraints. Identifying the similarities and differences associated with OSM-induced senescence versus EMT would help in the development of cancer-specific approaches to targeting the OSM/STAT3/SMAD3 axis.
Our studies support a previously reported cooperation between IL-6/STAT3 with TGF-β/SMAD3 signaling, mediated by both physical and functional complexes involving STAT3 and SMAD3 in hepatoma.Citation97 In addition, IL-6-mediated SMAD3 activation induces fibrosis in dermal fibroblasts, although via mechanisms that result in significantly elevated levels of SMAD3 phosphorylation.Citation98 This is distinct from our studies, where we see no increase in SMAD3 phosphorylation upon OSM exposure. Similarly, JAK/STAT3 activity is required for TGF-β-mediated Snail induction, EMT, and increased cell migration and invasion.Citation99 In fact, TGF-β itself has been reported to induce the phosphorylation of JAK1, STAT1, STAT3, and STAT5,Citation100 although this may be indirect, as TGF-β signaling induces IL-6 production, which can act in autocrine fashion to activate canonical JAK/STAT3 signaling.Citation101-106 Together, these studies support the cooperative intersection of activated-STAT3- and TGF-β-mediated signaling effectors. Yet, many other studies have reported disparate findings, suggesting that STAT3 activation and TGF-β signaling antagonize one another. For example, persistent STAT3 activation desensitizes cells to TGF-β signaling by increasing SMAD7 expression.Citation59 By depleting STAT3, TGF-β-mediated transcription could be induced and proliferation was suppressed. Moreover, as we describe, STAT3 was found in complex with SMAD3, but rather than increasing the transcription of SMAD3-regulated genes as observed in our study, the STAT3/SMAD3 interaction suppressed the SMAD3/SMAD4 interaction and prevented SMAD-mediated transcription.Citation107 Likewise, TGF-β signaling has been reported to downregulate IL-6-mediated STAT3 phosphorylation to prevent its nuclear translocation.Citation108,109 The differences underlying the cooperative vs. inhibitory nature of the STAT3/SMAD3 interactions is not yet understood, and supports the need for further study. One explanation is the number of different cells types (normal vs. transformed, epithelial vs. fibroblasts, immune cells, or keratinocytes) used in the cited studies. Not surprisingly, the response of different cell types to cytokine stimulation varies widely, depending on the genetic and epigenetic disposition of the exposed cells.
Finally, in addition to OSM, there are 11 different IL-6-family cytokines and at least 12 different co-receptors for gp130.Citation18,110-112 Beyond the IL-6 family, STAT3 can be activated by additional signaling cascades, many of which are also dysregulated in cancer. These include interferon (IFN) signaling, epidermal growth factor receptor (EGFR), bone marrow X-linked (BMX) non-receptor tyrosine kinase, SRC, and mutant PI3K.Citation113,114 In addition, the treatment of oncogene-addicted tumor cells treated with EGFR or MEK inhibitors activate STAT3 as a compensatory response, which can induce drug resistance.Citation115 We postulate that STAT3 activation by these additional means may also be able to drive mesenchymal/CSC properties via interconnection with TGF-β effector signaling. With regard to cancer therapies, suppression of STAT3 in combination with the targeted kinase inhibitors or chemotherapy could prevent the acquisition of more aggressive mesenchymal/CSC properties, and may therefore be a valuable addition to future therapeutic regimes. Yet, our studies suggest that suppressing STAT3 activation may have the unintended consequence of alleviating a tumor-suppressive senescence program, which may allow incipient cancers to resume proliferation. Future studies to identify key differences in (1) STAT3/SMAD3-mediated signaling responsible for the mesenchymal/CSC phenotype in cancer cells and (2) STAT3/SMAD3-mediated signaling responsible for the senescence program that inhibits proliferation in non-transformed cells would provide opportunities to refine therapeutic strategies and widen therapeutic windows.
Materials and methods
Cell culture conditions and reagents
Post-selection HMEC from specimen 48R, batch S (48RS-HMEC) with endogenous p16 promoter methylation were provided by Dr. Martha Stampfer (Lawrence Berkeley National Laboratory) and obtained from discarded surgical material of a reduction mammoplasty under Institutional Review Board (IRB) approval as previously described.Citation19,41,116 Non-immortalized 48RS-HMEC between passages 8 and 16 were grown in Medium 171 (Invitrogen, cat.# M171500, https://www.thermofisher.com/order/catalog/product/M171500) supplemented with mammary epithelial growth supplement (MEGS; Invitrogen, cat.# S0155, https://www.thermofisher.com/order/catalog/product/S0155) in a humidified atmosphere containing 5% CO2 at 37°C as described previously.Citation42,71 Phoenix–Ampho (ATCC, cat. # CRL-3213, https://www.atcc.org/Global/Products/0/1/4/3/CRL-3213.aspx) and HEK 293T (ATCC, cat. # CRL-3216, https://www.atcc.org/Products/All/CRL-3216.aspx) cell lines were grown in DMEM media (Corning cat. # 10–013-CV, http://cellgro.com/dulbecco-s-modification-of-eagle-s-medium-dmem-16.html) supplemented with 5% and 10% fetal bovine serum (FBS; Atlanta Biologicals, cat. # S103–50, https://www.atlantabio.com/catalog/animal-and-human-sera/fetal-bovine-serum/fetal-bovine-serum–premium) respectively, in a humidified atmosphere containing 5% CO2 at 37°C. Cells were treated with Recombinant human Oncostatin M (OSM) [10 ng/mL] (Peprotech, cat. # 300–10, https://www.peprotech.com/en-US/Pages/Product/Recombinant_Human_Oncostatin_M_(227_a.a.)/300–10), recombinant human Transforming Growth Factor-β1 (TGF-β1) [5 ng/mL] (Peprotech, cat. # 100–21C, https://www.peprotech.com/en-US/Pages/Product/Recombinant_Human_TGF-%CE%B21_(CHO_cell_derived)/100–21C), and small molecule TGFβR1 inhibitor compounds, SB525334 [2 μM] (Selleck Chemical, cat. # S1476, http://www.selleckchem.com/products/SB-525334.html) and SB431542 [10 μM] (Calbiochem, cat. # 616461, https://www.emdmillipore.com/US/en/product/TGF-%CE%B2-RI-Kinase-Inhibitor-VI%2C-SB431542—CAS-301836–41–9—Calbiochem,EMD_BIO-616461) by directly adding to the medium during each feeding for indicated experiments.
Viral vectors and infection
Retroviral vectors encoding wild-type STAT3 (WT-STAT3) and dominant-negative STAT3-Y705F (DN-STAT3) in LNCX2 were created by subcloning WT-STAT3 and DN-STAT3 cDNA from pLPCX-WT-STAT3 and pLPCX-DN-STAT3 vectors obtained from Dr. George Stark (Cleveland Clinic Foundation). Empty LNCX2 vector was used as a control and the LNCX2-DN-TGFβR2, LNCX2-SMAD7, LNCX2-CA-CyclinD1/CDK2, and LNCX2-MYC retroviral vectors are described elsewhere.Citation41,117 Retroviral vectors encoding shRNAs in pRetroSUPER-Puro targeting SMAD2 (pRetroSUPER-shSMAD2-Puro; Addgene, plasmid # 15722, http://www.addgene.org/15722/), SMAD3 (pRetroSUPER-shSMAD3-Puro; Addgene, plasmid # 15726, http://www.addgene.org/15726/), or SMAD4 (pRetroSUPER-shSMAD4-Puro; Addgene, plasmid # 15727, http://www.addgene.org/15727/) were gifts from and deposited to Addgene by Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center); and the pRetroSUPER-shGFP-Puro control vector was a kind gift from Dr. Yosef Shiloh (Tel Aviv University). The pLPCX SMAD3 vector (Addgene, plasmid # 12638, http://www.addgene.org/12638/), was a gift from and deposited to Addgene by Dr. Rik Derynck (University of California at San Francisco). Retroviruses were produced by transfecting retroviral vectors into Phoenix–Ampho cells simultaneously with a packaging plasmid that encodes the MLV-gag-pol and env genes using Lipofectamine 2000 (Invitrogen, cat. # 11668019, https://www.thermofisher.com/order/catalog/product/11668019) as previously described.Citation19,41,117 Lentivirus encoding shRNA targeting p53 (shp53) were produced by transfecting the lentiviral vector LVTHM-shp53 into 293T cells simultaneously with the second-generation packaging constructs pCMV-dR8.74 and pMD2G, kindly provided by Dr. Didier Trono (University of Geneva), using Lipofectamine 2000 as described elsewhere.Citation19,41,117 Supernatant Media 171 supplemented with .1% AlbuMax Bovine Serum Albumin (BSA; Invitrogen, cat. # 11020021, https://www.thermofisher.com/order/catalog/product/11020021) and containing virus was collected at 24–48 hours, filtered using a 0.22 mm filter, and supplemented with polybrene (4 μg/mL; Santa Cruz Biotechnology, cat. # sc-134220, http://www.scbt.com/datasheet-134220-polybrene.html) prior to being used to infect cells for 48 hours in a humidified atmosphere containing 5% CO2 at 37°C. Uninfected cells were removed by selection using the appropriate antibiotic: G418 [200 mg/mL] (Sigma, cat. # 4727878001, http://www.sigmaaldrich.com/catalog/product/roche/g418ro), and puromycin [1 mg/mL] (Sigma, cat. # P8833, http://www.sigmaaldrich.com/catalog/product/sigma/p8833).
Relative growth assays and western blotting analysis
For relative growth assays, cells were plated in triplicate in 6-well plates at 25,000 cells/well and treated with reagents for 7 d for indicated experiments, then total cell number for each well was quantified using a Beckman Coulter counter as previously described.Citation19,41 Mean total cell number for each triplicate is shown; and each treated-cell triplicate was plated in duplicate to obtain enough cells to acquire protein samples that correspond with each respective growth assay. For western blotting analysis, whole-cell protein extracts were prepared by incubating cell pellets in NP-40 single-lysis buffer containing protease inhibitor cocktail (Sigma-Aldrich, cat. # P8340, http://www.sigmaaldrich.com/catalog/product/sigma/p8340) and phosphatase inhibitor cocktail (Roche, cat. # 04906845001); equal quantities of proteins for each cell extract (determined by the Bradford method; Bio-Rad, cat. # 500–0006, http://www.bio-rad.com/en-us/sku/5000006-bio-rad-protein-assay-dye-reagent-concentrate-450-ml?parentCategoryGUID = d4d4169a-12e8–4819–8b3e-ccab019c6e13) were separated by SDS-PAGE on precast 4%–20% acrylamide gels (Thermo-Scientific, cat. # WT4201BOX, https://www.thermofisher.com/order/catalog/product/WT4201BOX), transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, cat. # NF-IPVH00010, http://www.emdmillipore.com/US/en/product/Immobilon-P-Membrane%2C-PVDF%2C-0.45%C2%A0%C2%B5m%2C-26.5%C2%A0cm-x%C2%A03.75%C2%A0m-roll,MM_NF-IPVH00010), and immunoblotted with the indicated primary antibodies. Primary antibodies were obtained from Cell Signaling Technology to STAT3 (cat. # 9139, http://www.cellsignal.com/products/primary-antibodies/stat3–124h6-mouse-mab/9139), P-STAT3 (Tyr705) (cat. # 9145, http://www.cellsignal.com/products/primary-antibodies/phospho-stat3-tyr705-d3a7-xp-rabbit-mab/9145), SMAD2 (cat. # 5339, http://www.cellsignal.com/products/primary-antibodies/smad2-d43b4-xp-rabbit-mab/5339), P-SMAD2 (Ser465/467) (cat. # 3108, http://www.cellsignal.com/products/primary-antibodies/phospho-smad2-ser465–467–138d4-rabbit-mab/3108), SMAD3 (cat. # 9523, http://www.cellsignal.com/products/primary-antibodies/smad3-c67h9-rabbit-mab/9523), P-SMAD3 (Ser423/425) (cat. # 9520, http://www.cellsignal.com/products/primary-antibodies/phospho-smad3-ser423–425-c25a9-rabbit-mab/9520), SMAD4 (cat. # 9515), E-cadherin (cat. # 3195, http://www.cellsignal.com/products/primary-antibodies/e-cadherin-24e10-rabbit-mab/3195), and Vimentin (cat. # 5741, http://www.cellsignal.com/products/primary-antibodies/vimentin-d21h3-xp-rabbit-mab/5741); and primary antibodies to β-actin (cat. # MAB1501, https://www.emdmillipore.com/US/en/product/Anti-Actin-Antibody%2C-clone-C4,MM_NF-MAB1501) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cat.# CB1001, https://www.emdmillipore.com/US/en/product/Anti-GAPDH-Mouse-mAb-%286C5%29,EMD_BIO-CB1001) were purchased from Millipore and Calbiochem respectively. Primary antibodies were detected with goat anti-mouse (Cell Signaling Technology, cat. # 7076, http://www.cellsignal.com/products/secondary-antibodies/anti-mouse-igg-hrp-linked-antibody/7076) or goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase (Cell Signaling Technology, cat. # 7074, http://www.cellsignal.com/products/secondary-antibodies/anti-rabbit-igg-hrp-linked-antibody/7074) using enhanced chemiluminescence (Perkin-Elmer, cat. # NEL104001EA, http://www.perkinelmer.com/product/western-lightning-plus-ecl-340ml-nel104001ea) on HyBlot CL film (Denville Scientific, cat. # E3018, http://www.denvillescientific.com/node/1226).
Co-immunoprecipitation and subcellular protein fractionations
For co-immunoprecipitation studies, HEK 293T cells were transiently transfected with 12 μg pLPCX-SMAD3 in combination with 12 μg pLPCX-WT-STAT3 or pLPCX-DN-STAT3 Y705F using Lipofectamine 2000. After transfection for 24 hours at 37°C in a humidified chamber containing 5% CO2, transfected cells were split into 2 samples: one sample of each cell derivative was left untreated while the second sample was treated 2 d with recombinant OSM. Cells were washed and harvested using ice-cold PBS and fixed in 4% formaldehyde-PBS for 10 minutes at room temperature. Formaldehyde cross-linking was neutralized using 100 mM glycine-PBS for 1 minute at room temperature and cells were lysed in RIPA lysis buffer containing protease inhibitor cocktail and phosphatase inhibitor cocktail. Cell lysates were then sonicated for 5 minutes at 4°C with a cycle of 30 seconds on/30 seconds off and then placed on ice for 30 minutes. One milligram of each lysate was pre-cleared with protein G Dynabeads (Invitrogen, cat.# 10004D, https://www.thermofisher.com/order/catalog/product/10004D) for 1 hour at 4°C with constant rotation; 50 μg of each sample was also extracted and combined with BME-SDS loading dye as a 5% input-sample for western blotting analysis. Pre-cleared samples were centrifuged and the supernatants were transferred to new tubes, then subjected to immunoprecipitation by incubating at 4°C for 3 hours with 10 μg of primary anti-SMAD3 conjugated to protein G Dynabeads and constant rotation. After incubation, samples were centrifuged for 1 minute at 10,000 rpm, supernatants discarded, and remaining agarose beads were washed 6 times with RIPA lysis buffer and combined with BME-SDS loading dye. Immunoprecipitations and 1% Inputs were heated at 100°C for 15 minutes, then separated by SDS–PAGE, transferred to PVDF membrane, and subjected to western analysis following blocking with 5% BSA/PBS-T buffer. For subcellular protein localization assays, cells were treated for 4 d with OSM or TGF-β1 alone and subjected to a subcellular protein fractionation kit (Thermo-Scientific, cat. #78840, https://www.thermofisher.com/order/catalog/product/78840) performed according to the manufacturer's instructions. Protein extracts from the nuclear-soluble fractions were analyzed by western blot to examine nuclear localization of STAT3 and SMAD proteins.
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total messenger ribonucleic acid (mRNA) was harvested from cells using an RNeasy Kit (Qiagen, cat. # 74104, https://www.qiagen.com/us/shop/sample-technologies/rna/rna-preparation/rneasy-mini-kit/#orderinginformation) with on-column DNase I (Qiagen, cat. # 79254, https://www.qiagen.com/us/shop/sample-technologies/rna/rna-preparation/rneasy-mini-kit/RNase-Free-DNase-Set#orderinginformation) digest, then mRNA (1 μg) was reverse transcribed to make cDNA using the iScript cDNA synthesis Kit (Bio-Rad, cat. # 170–8891, http://www.bio-rad.com/en-us/sku/1708891-iscript-cdna-synthesis-kit-100-x-20-ul-rxns?parentCategoryGUID = M87EWZESH), and the cDNA subjected to qRT-PCR using iQ SYBR Green Supermix (Bio-Rad, cat. # 170–8882, http://www.bio-rad.com/en-us/sku/1708882-iq-sybr-green-supermix-500-x-50-ul-rxns-12–5-ml-10-x-1–25-ml?parentCategoryGUID = M7FBG34VY). The RTCitation2 Profiler qRT-PCR Array System for either TGF-β Signaling Targets (SA Biosciences, cat. # PAHS-235Z, http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-235Z.html) in 2D-grown cells, or for Extracellular Matrix (ECM) and Adhesion Molecules (SA Biosciences, cat. # PAHS-0135Z, http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-013Z.html) in 3D-matrigel grown cells, was analyzed on a CFX96 thermocycler (Bio-Rad) according to the manufacturer's instructions. For single-gene qRT-PCR analysis, the following primer sequences (listed 5’-to-3’) were designed and used to amplify genes with a 60°C annealing temp: CDH1 Forward CCCAATACATCTCCCTTCACAG; CDH1 Reverse CCACCTCTAAGGCCATCTTTG; SNAI1 Forward GGAAGCCTAACTACAGCGAG; SNAI1 Reverse CAGAGTCCCAGATGAGCATTG; TNFSF10 Forward GAGCTGAAGCAGATGCAGGAC; TNFSF10 Reverse TGACGGAGTTGCCACTTGACT; MYC Forward CAGCTGCTTAGACGCTGGATT; MYC Reverse GTAGAAATACGGCTGCACCGA; ATF3 Forward AAGAACGAGAAGCAGCATTTGA; ATF3 Reverse TTCTGAGCCCGGACAATACAC; ACTIN Forward CAGCCATGTACGTTGCTATCCAGG; ACTIN Reverse AGGTCCAGACGCAGGATGGCATG.
3-Dimensional (3D) cultures and microscopy
For AIG assays, soft agar was made by mixing 2X type VII agarose (Sigma-Aldrich, cat. # A4018, http://www.sigmaaldrich.com/catalog/product/sial/a0701) with 2X Mammary Epithelial Basal medium MCDB170 (US Biologicals, cat. # M2162, https://www.usbio.net/media/M2162) supplemented with MEGS, o-phosphoethanolamine [0.1 mmol/L] (Sigma, cat. # P0503, http://www.sigmaaldrich.com/catalog/product/sigma/p0503), and ethanolamine [0.1 mmol/L] (Sigma, cat. # E0135, http://www.sigmaaldrich.com/catalog/product/sial/e0135) as previously described.Citation19,41,117 Cells (1 × 105) were counted using a Beckman Coulter counter, suspended in 0.6% soft agar, and plated onto a bottom layer of 1.2% soft agar in 60-mm plates in triplicate, as described previously.Citation19,41,117 Cells were cultured for 2 weeks, with the medium and respective cytokine treatments for indicated experiments being changed every 3 d. AIG was quantified by scanning each plate with an auto-mated multipanel scanning microscope, and analyzing the stitched digital images using MetaMorph software (Molecular Devices) to acquire soft agar colony counts for each entire plate. Graphs representing the mean of at least 3 counts for each experiment and error bars representing the calculated standard deviation from the mean of the 3 independent plate counts were produced in Excel (Microsoft). For 3-dimensional (3D) invasion assays, 1×105 cells were counted using a Beckman Coulter counter and then suspended in growth factor–reduced, laminin-rich basement membrane (Matrigel; Corning, cat. # 354230, http://www.corning.com/worldwide/en/products/life-sciences/products/surfaces/matrigel-matrix.html) for each well in 6-well plates. Cells were cultured for 2 weeks, with the medium and respective cytokine treatments for indicated experiments being changed every 3 d. The development of an invasive phenotype was visually characterized by confocal and bright field microscopy at 5X magnification. For confocal Immunofluorescence analysis of 3D cultures, 1 × 105 cells were plated onto 35-mm glass bottom dishes (MatTek Corp., cat. # P35G-1.5–20-C, https://www.mattek.com/store/p35g-0–170–14-c/) in 10% top-layer matrigel using the 3D on-top assay as described.Citation118 Cells were cultured 1 week with medium and respective cytokine treatments for indicated experiments exchanged every 2 d. Cells were washed with ice-cold PBS, fixed and permeabilized in ice-cold 100% methanol for 15 minutes, blocked with 5% BSA-Immunofluorescence (IF) buffer (0.2% Triton X-100, 0.1% BSA, 0.05% Tween 20 in PBS), and stained overnight with the primary antibodies, anti-E-cadherin conjugated to Alexafluor 647 (Santa Cruz Biotechnology, cat. # sc-21791 AF647, http://www.scbt.com/datasheet-21791-e-cadherin-67a4-antibody.html) and anti-Vimentin conjugated to Alexafluor 488 (Santa Cruz Biotechnology, cat. # sc-6260 AF488, http://www.scbt.com/datasheet-6260-vimentin-v9-antibody.html), at 1:50 dilutions in IF buffer; nuclei were counterstained with Diaminophenylindole (DAPI) [1 μg/mL] (Sigma-Aldrich, cat. # D9542, http://www.sigmaaldrich.com/catalog/product/sigma/d9542) for 5 minutes immediately prior to confocal microscopy analysis. Confocal images were captured using a ×63 oil immersion objective on a Zeiss LSM 510 multiphoton microscope and AIM software (Leica). All bright-field images were obtained at 5X magnification with a Leica DMI6000 microscope and then analyzed with Metamorph software to quantify the development of an invasive phenotype. Percent of acini formation/total acini invasion was calculated using the formular: [# Invasive Acini/# Total Acini] x100.
Abbreviations
| AIG | = | anchorage independent growth |
| BMX | = | bone marrow X-linked. |
| CSC | = | cancer stem cell |
| ECM | = | extracellular matrix |
| EGFR | = | epidermal growth factor receptor |
| EMT | = | epithelial-mesenchymal transition |
| gp130 | = | glycoprotein 130 |
| HMEC | = | human mammary epithelial cells |
| IFN | = | interferon |
| IL-6 | = | Interleukin-6 |
| JAK | = | Janus Kinase |
| MAPK | = | mitogen-activated protein kinase |
| OIS | = | oncogene-induced senescence |
| OSM | = | Oncostatin M |
| OSMRβ | = | OSM receptor subunit β |
| PI3K | = | phosphatidylinositol 3-kinase |
| SBE | = | SMAD-binding elements |
| STAT3 | = | Signal Transducer and Activator of Transcription 3 |
| TGF-β | = | Transforming Growth Factor-β |
| TME | = | tumor microenvironment |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the US National Institutes of Health (R01CA138421 to M.W.J.; T32CA059366 to R.C. and D.J.J.) and the American Cancer Society (Research Scholar Award # RSG CCG-122517). Additional support was provided by the Case Comprehensive Cancer Center (P30 CA43703; Cytometry & Imaging Microscopy Core Facility).
References
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7:41-51; PMID:17186030; http://dx.doi.org/10.1038/nri1995
- Culig Z. Cytokine disbalance in common human cancers. Biochim Biophys Acta 2011; 1813:308-14; PMID:21167870; http://dx.doi.org/10.1016/j.bbamcr.2010.12.010
- Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008; 43:374-9; PMID:18701317; http://dx.doi.org/10.1016/j.cyto.2008.07.014
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 2007; 117:1175-83; PMID:17476347; http://dx.doi.org/10.1172/JCI31537
- Blanchard F, Duplomb L, Baud'huin M, Brounais B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev 2009; 20:19-28; PMID:19038573; http://dx.doi.org/10.1016/j.cytogfr.2008.11.004
- Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 2011; 22:83-9; PMID:21377916; http://dx.doi.org/10.1016/j.cytogfr.2011.02.003
- Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013; 15:848-62; PMID:23814496; http://dx.doi.org/10.1593/neo.13706
- Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAKSTAT 2013; 2:e25763; PMID:24416650
- Chevalier S, Fourcin M, Robledo O, Wijdenes J, Pouplard-Barthelaix A, Gascan H. Interleukin-6 family of cytokines induced activation of different functional sites expressed by gp130 transducing protein. J Biol Chem 1996; 271:14764-72; PMID:8662918; http://dx.doi.org/10.1074/jbc.271.25.14764
- Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38:904-10; PMID:22651903; http://dx.doi.org/10.1016/j.ctrv.2012.04.007
- Tanaka M, Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol 2003; 149:39-52; PMID:12811586
- Hermanns HM, Radtke S, Haan C, Schmitz-Van de Leur H, Tavernier J, Heinrich PC, Behrmann I. Contributions of leukemia inhibitory factor receptor and oncostatin M receptor to signal transduction in heterodimeric complexes with glycoprotein 130. J Immunol 1999; 163:6651-8; PMID:10586060
- Tiffen PG, Omidvar N, Marquez-Almuina N, Croston D, Watson CJ, Clarkson RW. A dual role for oncostatin M signaling in the differentiation and death of mammary epithelial cells in vivo. Mol Endocrinol 2008; 22:2677-88; PMID:18927239; http://dx.doi.org/10.1210/me.2008-0097
- Grant SL, Begley CG. The oncostatin M signalling pathway: reversing the neoplastic phenotype? Mol Med Today 1999; 5:406-12; PMID:10462753; http://dx.doi.org/10.1016/S1357-4310(99)01540-3
- Anhuf D, Weissenbach M, Schmitz J, Sobota R, Hermanns HM, Radtke S, Linnemann S, Behrmann I, Heinrich PC, Schaper F. Signal transduction of IL-6, leukemia-inhibitory factor, and oncostatin M: structural receptor requirements for signal attenuation. J Immunol 2000; 165:2535-43; PMID:10946280; http://dx.doi.org/10.4049/jimmunol.165.5.2535
- Dey G, Radhakrishnan A, Syed N, Thomas JK, Nadig A, Srikumar K, Mathur PP, Pandey A, Lin SK, Raju R, et al. Signaling network of Oncostatin M pathway. J Cell Commun Signal 2013; 7:103-8; PMID:23255051; http://dx.doi.org/10.1007/s12079-012-0186-y
- Auguste P, Guillet C, Fourcin M, Olivier C, Veziers J, Pouplard-Barthelaix A, Gascan H. Signaling of type II oncostatin M receptor. J Biol Chem 1997; 272:15760-4; PMID:9188471; http://dx.doi.org/10.1074/jbc.272.25.15760
- Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol 2010; 88:1145-56; PMID:20610800; http://dx.doi.org/10.1189/jlb.0410217
- Kan CE, Cipriano R, Jackson MW. c-MYC functions as a molecular switch to alter the response of human mammary epithelial cells to oncostatin M. Cancer Res 2011; 71:6930-9; PMID:21975934; http://dx.doi.org/10.1158/0008-5472.CAN-10-3860
- West NR, Murphy LC, Watson PH. Oncostatin M suppresses oestrogen receptor-alpha expression and is associated with poor outcome in human breast cancer. Endocr Relat Cancer 2012; 19:181-95; PMID:22267707; http://dx.doi.org/10.1530/ERC-11-0326
- West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014; 33:1485-94; PMID:23584474; http://dx.doi.org/10.1038/onc.2013.105
- Vlaicu P, Mertins P, Mayr T, Widschwendter P, Ataseven B, Hogel B, Eiermann W, Knyazev P, Ullrich A. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC Cancer 2013; 13:197; PMID:23597096; http://dx.doi.org/10.1186/1471-2407-13-197
- Ryan RE, Martin B, Mellor L, Jacob RB, Tawara K, McDougal OM, Oxford JT, Jorcyk CL. Oncostatin M binds to extracellular matrix in a bioactive conformation: implications for inflammation and metastasis. Cytokine 2015; 72:71-85; PMID:25622278; http://dx.doi.org/10.1016/j.cyto.2014.11.007
- Zhu M, Che Q, Liao Y, Wang H, Wang J, Chen Z, Wang F, Dai C, Wan X. Oncostatin M activates STAT3 to promote endometrial cancer invasion and angiogenesis. Oncol Rep 2015; 34:129-38; PMID:25954856
- Guo L, Chen C, Shi M, Wang F, Chen X, Diao D, Hu M, Yu M, Qian L, Guo N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013; 32:5272-82; PMID:23318420; http://dx.doi.org/10.1038/onc.2012.573
- Underhill-Day N, Heath JK. Oncostatin M (OSM) cytostasis of breast tumor cells: characterization of an OSM receptor beta-specific kernel. Cancer Res 2006; 66:10891-901; PMID:17108126; http://dx.doi.org/10.1158/0008-5472.CAN-06-1766
- Hutt JA, DeWille JW. Oncostatin M induces growth arrest of mammary epithelium via a CCAAT/enhancer-binding protein delta-dependent pathway. Mol Cancer Ther 2002; 1:601-10; PMID:12479220
- Holzer RG, Ryan RE, Tommack M, Schlekeway E, Jorcyk CL. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: role of cyclooxygenase-2. Clin Exp Metastasis 2004; 21:167-76; PMID:15168734; http://dx.doi.org/10.1023/B:CLIN.0000024760.02667.db
- Crichton MB, Nichols JE, Zhao Y, Bulun SE, Simpson ER. Expression of transcripts of interleukin-6 and related cytokines by human breast tumors, breast cancer cells, and adipose stromal cells. Mol Cell Endocrinol 1996; 118:215-20; PMID:8735608; http://dx.doi.org/10.1016/0303-7207(96)03761-6
- Douglas AM, Grant SL, Goss GA, Clouston DR, Sutherland RL, Begley CG. Oncostatin M induces the differentiation of breast cancer cells. Int J Cancer 1998; 75:64-73; PMID:9426692; http://dx.doi.org/10.1002/(SICI)1097-0215(19980105)75:1%3c64::AID-IJC11%3e3.0.CO;2-D
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008; 14:518-27; PMID:18438415; http://dx.doi.org/10.1038/nm1764
- Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. OSM, LIF, its receptors, and its relationship with the malignance in human breast carcinoma (in situ and in infiltrative). Cancer Invest 2008; 26:222-9; PMID:18317962; http://dx.doi.org/10.1080/07357900701638491
- Jorcyk CL, Holzer RG, Ryan RE. Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine 2006; 33:323-36; PMID:16713283; http://dx.doi.org/10.1016/j.cyto.2006.03.004
- Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, Van Den Broecke R, Limame R, Mestdagh P, Vandesompele J, Vanhove C, et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res 2014; 74:6806-19; PMID:25252914; http://dx.doi.org/10.1158/0008-5472.CAN-14-0160
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012; 4:159-65; PMID:22394614; http://dx.doi.org/10.18632/aging.100443
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A 2014; 111:8832-7; PMID:24889617; http://dx.doi.org/10.1073/pnas.1405723111
- Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle 2014; 13:3628-35; PMID:25483060; http://dx.doi.org/10.4161/15384101.2014.985507
- Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene 2008; 27:2801-9; PMID:18193093; http://dx.doi.org/10.1038/sj.onc.1210950
- Liu J, Spence MJ, Wallace PM, Forcier K, Hellstrom I, Vestal RE. Oncostatin M-specific receptor mediates inhibition of breast cancer cell growth and down-regulation of the c-myc proto-oncogene. Cell Growth Differ 1997; 8:667-76; PMID:9186000
- Spence MJ, Vestal RE, Liu J. Oncostatin M-mediated transcriptional suppression of the c-myc gene in breast cancer cells. Cancer Res 1997; 57:2223-8; PMID:9187125
- Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci U S A 2011; 108:8668-73; PMID:21555587; http://dx.doi.org/10.1073/pnas.1015022108
- Olsen CL, Gardie B, Yaswen P, Stampfer MR. Raf-1-induced growth arrest in human mammary epithelial cells is p16-independent and is overcome in immortal cells during conversion. Oncogene 2002; 21:6328-39; PMID:12214273; http://dx.doi.org/10.1038/sj.onc.1205780
- Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 2009; 21:166-76; PMID:19237272; http://dx.doi.org/10.1016/j.ceb.2009.01.021
- Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol 2006; 16:313-7; PMID:16935001; http://dx.doi.org/10.1016/j.semcancer.2006.07.012
- Arvanitis C, Felsher DW. Conditionally MYC: insights from novel transgenic models. Cancer Lett 2005; 226:95-9; PMID:16039948; http://dx.doi.org/10.1016/j.canlet.2004.10.043
- Hynes NE, Stoelzle T. Key signalling nodes in mammary gland development and cancer: Myc. Breast Cancer Res 2009; 11:210; PMID:19849814; http://dx.doi.org/10.1186/bcr2406
- Pelengaris S, Khan M. The many faces of c-MYC. Arch Biochem Biophys 2003; 416:129-36; PMID:12893289; http://dx.doi.org/10.1016/S0003-9861(03)00294-7
- Xu J, Chen Y, Olopade OI. MYC and Breast Cancer. Genes Cancer 2010; 1:629-40; PMID:21779462; http://dx.doi.org/10.1177/1947601910378691
- Wolfer A, Ramaswamy S. MYC and metastasis. Cancer Res 2011; 71:2034-7; PMID:21406394; http://dx.doi.org/10.1158/0008-5472.CAN-10-3776
- Singhi AD, Cimino-Mathews A, Jenkins RB, Lan F, Fink SR, Nassar H, Vang R, Fetting JH, Hicks J, Sukumar S, et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol 2012; 25:378-87; PMID:22056952; http://dx.doi.org/10.1038/modpathol.2011.171
- Chen Y, Olopade OI. MYC in breast tumor progression. Expert Rev Anticancer Ther 2008; 8:1689-98; PMID:18925859; http://dx.doi.org/10.1586/14737140.8.10.1689
- Albihn A, Johnsen JI, Henriksson MA. MYC in Oncogenesis and as a Target for Cancer Therapies. Advances in Cancer Research 2010; 107:163-224; http://dx.doi.org/10.1016/S0065-230X(10)07006-5
- Dang CV. MYC on the path to cancer. Cell 2012; 149:22-35; PMID:22464321; http://dx.doi.org/10.1016/j.cell.2012.03.003
- Dang CV. MYC, microRNAs and glutamine addiction in cancers. Cell Cycle 2009; 8:3243-5; PMID:19806017; http://dx.doi.org/10.4161/cc.8.20.9522
- Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res 2010; 316:1429-37; PMID:20382143; http://dx.doi.org/10.1016/j.yexcr.2010.03.025
- Zhang F, Li C, Halfter H, Liu J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene 2003; 22:894-905; PMID:12584569; http://dx.doi.org/10.1038/sj.onc.1206158
- Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-beta-mediated induction of the CDK inhibitor p15(Ink4B). Mol Cell 2002; 9:133-43; PMID:11804592; http://dx.doi.org/10.1016/S1097-2765(01)00430-0
- Chen CRK, Y, Siegel, PM, Massagué, J E2F4/5 and p107 as Smad Cofactors Linking the TGFβ Receptor to c-myc Repression. Cell 2002; 110:19-32; PMID:12150994; http://dx.doi.org/10.1016/S0092-8674(02)00801-2
- Luwor RB, Baradaran B, Taylor LE, Iaria J, Nheu TV, Amiry N, Hovens CM, Wang B, Kaye AH, Zhu HJ. Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene 2013; 32:2433-41; PMID:22751114; http://dx.doi.org/10.1038/onc.2012.260
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24:2463-79; PMID:21078816; http://dx.doi.org/10.1101/gad.1971610
- Moir JA, White SA, Mann J. Arrested development and the great escape–the role of cellular senescence in pancreatic cancer. Int J Biochem Cell Biol 2014; 57:142-8; PMID:25461770; http://dx.doi.org/10.1016/j.biocel.2014.10.018
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends in Cell Biol 2001; 11:S27-S31; PMID:11684439; http://dx.doi.org/10.1016/S0962-8924(01)82148-6
- Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol 2008; 20:150-5; PMID:18353625; http://dx.doi.org/10.1016/j.ceb.2008.01.007
- Evan GI, d'Adda di Fagagna F. Cellular senescence: hot or what? Curr Opin Genet Dev 2009; 19:25-31; PMID:19181515; http://dx.doi.org/10.1016/j.gde.2008.11.009
- Saab R. Cellular senescence: many roads, one final destination. ScientificWorldJournal 2010; 10:727-41; PMID:20419281; http://dx.doi.org/10.1100/tsw.2010.68
- Larsson LG. Cellular senescence–a barrier against tumor development? Semin Cancer Biol 2011; 21:347-8; PMID:22137673; http://dx.doi.org/10.1016/j.semcancer.2011.11.001
- Saab R. Senescence and pre-malignancy: how do tumors progress? Semin Cancer Biol 2011; 21:385-91; PMID:21982725; http://dx.doi.org/10.1016/j.semcancer.2011.09.013
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10:51-7; PMID:20029423; http://dx.doi.org/10.1038/nrc2772
- DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer 2009; 45 Suppl 1:211-6; PMID:19775620; http://dx.doi.org/10.1016/S0959-8049(09)70036-X
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 2006; 6:24-37; PMID:16397525; http://dx.doi.org/10.1038/nrc1782
- Levano KS, Jung EH, Kenny PA. Breast cancer subtypes express distinct receptor repertoires for tumor-associated macrophage derived cytokines. Biochem Biophys Res Commun 2011; 411:107-10; PMID:21712030; http://dx.doi.org/10.1016/j.bbrc.2011.06.102
- Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. Mass-spectrometry-based draft of the human proteome. Nature 2014; 509:582-7; PMID:24870543; http://dx.doi.org/10.1038/nature13319
- Grenier A, Dehoux M, Boutten A, Arce-Vicioso M, Durand G, Gougerot-Pocidalo MA, Chollet-Martin S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 1999; 93:1413-21; PMID:9949186
- Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, Mao F, Wang M, Zhu W, Qian H, et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis 2014; 5:e1295; PMID:24946088; http://dx.doi.org/10.1038/cddis.2014.263
- Suda T, Chida K, Todate A, Ide K, Asada K, Nakamura Y, Suzuki K, Kuwata H, Nakamura H. Oncostatin M production by human dendritic cells in response to bacterial products. Cytokine 2002; 17:335-40; PMID:12061841; http://dx.doi.org/10.1006/cyto.2002.1023
- Hermanns HM, Radtke S, Schaper F, Heinrich PC, Behrmann I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J Biol Chem 2000; 275:40742-8; PMID:11016927; http://dx.doi.org/10.1074/jbc.M005408200
- Hermanns HM. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015; 26:545-58; PMID:26198770; http://dx.doi.org/10.1016/j.cytogfr.2015.07.006
- Richards CD. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm 2013; 2013:512103; PMID:24381786; http://dx.doi.org/10.1155/2013/512103
- Kretzschmar M. Transforming growth factor-beta and breast cancer - Transforming growth factor-beta/SMAD signaling defects and cancer. Breast Cancer Res 2000; 2:107-15; PMID:11250700; http://dx.doi.org/10.1186/bcr42
- Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 2005; 65:8896-904; PMID:16204061; http://dx.doi.org/10.1158/0008-5472.CAN-05-1734
- Mohamed MM, El-Ghonaimy EA, Nouh MA, Schneider RJ, Sloane BF, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int J Biochem Cell Biol 2014; 46:138-47; PMID:24291763; http://dx.doi.org/10.1016/j.biocel.2013.11.015
- Hoermann G, Cerny-Reiterer S, Herrmann H, Blatt K, Bilban M, Gisslinger H, Gisslinger B, Mullauer L, Kralovics R, Mannhalter C, et al. Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J 2012; 26:894-906; PMID:22051730; http://dx.doi.org/10.1096/fj.11-193078
- Royuela M, Ricote M, Parsons MS, Garcia-Tunon I, Paniagua R, de Miguel MP. Immunohistochemical analysis of the IL-6 family of cytokines and their receptors in benign, hyperplasic, and malignant human prostate. J Pathol 2004; 202:41-9; PMID:14694520; http://dx.doi.org/10.1002/path.1476
- Rychli K, Kaun C, Hohensinner PJ, Rega G, Pfaffenberger S, Vyskocil E, Breuss JM, Furnkranz A, Uhrin P, Zaujec J, et al. The inflammatory mediator oncostatin M induces angiopoietin 2 expression in endothelial cells in vitro and in vivo. J Thromb Haemost 2010; 8:596-604; PMID:20088942; http://dx.doi.org/10.1111/j.1538-7836.2010.03741.x
- Lee MJ, Heo SC, Shin SH, Kwon YW, Do EK, Suh DS, Yoon MS, Kim JH. Oncostatin M promotes mesenchymal stem cell-stimulated tumor growth through a paracrine mechanism involving periostin and TGFBI. Int J Biochem Cell Biol 2013; 45:1869-77; PMID:23735324; http://dx.doi.org/10.1016/j.biocel.2013.05.027
- Li Q, Zhu J, Sun F, Liu L, Liu X, Yue Y. Oncostatin M promotes proliferation of ovarian cancer cells through signal transducer and activator of transcription 3. Int J Mol Med 2011; 28:101-8; PMID:21399864
- Lauber S, Wong S, Cutz JC, Tanaka M, Barra N, Lhotak S, Ashkar A, Richards CD. Novel function of Oncostatin M as a potent tumour-promoting agent in lung. Int J Cancer 2015; 136:831-43; PMID:24976180; http://dx.doi.org/10.1002/ijc.29055
- Ng G, Winder D, Muralidhar B, Gooding E, Roberts I, Pett M, Mukherjee G, Huang J, Coleman N. Gain and overexpression of the oncostatin M receptor occur frequently in cervical squamous cell carcinoma and are associated with adverse clinical outcome. J Pathol 2007; 212:325-34; PMID:17516585; http://dx.doi.org/10.1002/path.2184
- Smith DA, Kiba A, Zong Y, Witte ON. Interleukin-6 and oncostatin-M synergize with the PI3K/AKT pathway to promote aggressive prostate malignancy in mouse and human tissues. Mol Cancer Res 2013; 11:1159-65; PMID:23867565; http://dx.doi.org/10.1158/1541-7786.MCR-13-0238
- Torres C, Perales S, Alejandre MJ, Iglesias J, Palomino RJ, Martin M, Caba O, Prados JC, Aranega A, Delgado JR, et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 2014; 43:1042-9; PMID:24979617; http://dx.doi.org/10.1097/MPA.0000000000000155
- Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt ML, Bhadauria S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5:5350-68; PMID:25051364; http://dx.doi.org/10.18632/oncotarget.2110
- Winder DM, Chattopadhyay A, Muralidhar B, Bauer J, English WR, Zhang X, Karagavriilidou K, Roberts I, Pett MR, Murphy G, et al. Overexpression of the oncostatin M receptor in cervical squamous cell carcinoma cells is associated with a pro-angiogenic phenotype and increased cell motility and invasiveness. J Pathol 2011; 225:448-62; PMID:21952923; http://dx.doi.org/10.1002/path.2968
- Gurluler E TL, Guner OS, Kucukmetin NT, Hizli B, Zorluoglu A. Oncostatin-M as a novel biomarker in colon cancer patients and its association with clinicopathologic variables. Eur Rev Med Pharmacol Sci 2014; 18:2042-7; PMID:25027345
- Caffarel MM, Coleman N. Oncostatin M receptor is a novel therapeutic target in cervical squamous cell carcinoma. J Pathol 2014; 232:386-90; PMID:24659184; http://dx.doi.org/10.1002/path.4305
- Singh RA, Sodhi A. Cisplatin-treated macrophages produce oncostatin M: regulation by serine/threonine and protein tyrosine kinases/phosphatases and Ca2+/calmodulin. Immunol Lett 1998; 62:159-64; PMID:9698114; http://dx.doi.org/10.1016/S0165-2478(98)00040-6
- Sodhi A, Shishodia S, Shrivastava A. Cisplatin-stimulated murine bone marrow-derived macrophages secrete oncostatin M. Immunol Cell Biol 1997; 75:492-6; PMID:9429898; http://dx.doi.org/10.1038/icb.1997.76
- Yamamoto T, Matsuda T, Muraguchi A, Miyazono K, Kawabata M. Cross-talk between IL-6 and TGF-beta signaling in hepatoma cells. Febs Letters 2001; 492:247-53; PMID:11257503; http://dx.doi.org/10.1016/S0014-5793(01)02258-X
- O'Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem 2014; 289:9952-60; PMID:24550394; http://dx.doi.org/10.1074/jbc.M113.545822
- Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT. JAK/STAT3 signaling is required for TGF-beta-induced epithelial-mesenchymal transition in lung cancer cells. Int J Oncol 2014; 44:1643-51; PMID:24573038
- Dong Z, Tai W, Lei W, Wang Y, Li Z, Zhang T. IL-27 inhibits the TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. BMC Cell Biol 2016; 17:7; PMID:26932661; http://dx.doi.org/10.1186/s12860-016-0084-x
- Moon SU, Kang MH, Sung JH, Kim JW, Lee JO, Kim YJ, Lee KW, Bang SM, Lee JS, Kim JH. Effect of Smad3/4 on chemotherapeutic drug sensitivity in colorectal cancer cells. Oncol Rep 2015; 33:185-92; PMID:25370208
- Turner M, Chantry D, Feldmann M. Transforming growth factor beta induces the production of interleukin 6 by human peripheral blood mononuclear cells. Cytokine 1990; 2:211-6; PMID:2104224; http://dx.doi.org/10.1016/1043-4666(90)90018-O
- Seong GJ, Hong S, Jung SA, Lee JJ, Lim E, Kim SJ, Lee JH. TGF-beta-induced interleukin-6 participates in transdifferentiation of human Tenon's fibroblasts to myofibroblasts. Mol Vis 2009; 15:2123-8; PMID:19862334
- Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Roth M. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem 1999; 274:12933-8; PMID:10212284; http://dx.doi.org/10.1074/jbc.274.18.12933
- Elias JA, Lentz V, Cummings PJ. Transforming growth factor-beta regulation of IL-6 production by unstimulated and IL-1-stimulated human fibroblasts. J Immunol 1991; 146:3437-43; PMID:2026873
- Park JI, Lee MG, Cho K, Park BJ, Chae KS, Byun DS, Ryu BK, Park YK, Chi SG. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene 2003; 22:4314-32; PMID:12853969; http://dx.doi.org/10.1038/sj.onc.1206478
- Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, Lin X, Feng XH. STAT3 selectively interacts with Smad3 to antagonize TGF-beta. Oncogene 2016; 35:4388-98; PMID:26616859; http://dx.doi.org/10.1038/onc.2015.446
- Zhao S, Venkatasubbarao K, Lazor JW, Sperry J, Jin C, Cao L, Freeman JW. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor beta-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res 2008; 68:4221-8; PMID:18519681; http://dx.doi.org/10.1158/0008-5472.CAN-07-5123
- Walia B, Wang L, Merlin D, Sitaraman SV. TGF-beta down-regulates IL-6 signaling in intestinal epithelial cells: critical role of SMAD-2. FASEB J 2003; 17:2130-2; PMID:14500551
- Muller-Newen G. The cytokine receptor gp130: faithfully promiscuous. Sci STKE 2003; 2003:PE40; PMID:14506288
- Suthaus J, Tillmann A, Lorenzen I, Bulanova E, Rose-John S, Scheller J. Forced homo- and heterodimerization of all gp130-type receptor complexes leads to constitutive ligand-independent signaling and cytokine-independent growth. Mol Biol Cell 2010; 21:2797-807; PMID:20554759; http://dx.doi.org/10.1091/mbc.E10-03-0240
- Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood 1995; 86:1243-54; PMID:7632928
- Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, MacSwords J, Eyler CE, McLendon RE, Heddleston JM, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011; 19:498-511; PMID:21481791; http://dx.doi.org/10.1016/j.ccr.2011.03.004
- Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol 1998; 18:2545-52; PMID:9566874; http://dx.doi.org/10.1128/MCB.18.5.2545
- Vultur A, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, Li L, Gimotty PA, Wilson M, Hayden J, et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene 2014; 33:1850-61; PMID:23624919; http://dx.doi.org/10.1038/onc.2013.131
- Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res 2009; 69:7557-68; PMID:19773443; http://dx.doi.org/10.1158/0008-5472.CAN-09-0270
- Junk DJ, Cipriano R, Stampfer M, Jackson MW. Constitutive CCND1/CDK2 activity substitutes for p53 loss, or MYC or oncogenic RAS expression in the transformation of human mammary epithelial cells. PLoS One 2013; 8:e53776; PMID:23390492; http://dx.doi.org/10.1371/journal.pone.0053776
- Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 2007; 4:359-65; PMID:17396127; http://dx.doi.org/10.1038/nmeth1015