
ABSTRACT
Proteins essential for homologous recombination play a pivotal role in the repair of DNA double strand breaks, DNA inter-strand crosslinks and replication fork stability. Defects in homologous recombination also play a critical role in the development of cancer and the sensitivity of these cancers to chemotherapy. RAD51, an essential factor for homologous recombination and replication fork protection, accumulates and forms immunocytochemically detectable nuclear foci at sites of DNA damage. To identify kinases that may regulate RAD51 localization to sites of DNA damage, we performed a human kinome siRNA library screen, using DNA damage-induced RAD51 foci formation as readout. We found that NEK8, a NIMA family kinase member, is required for efficient DNA damage-induced RAD51 foci formation. Interestingly, knockout of Nek8 in murine embryonic fibroblasts led to cellular sensitivity to the replication inhibitor, hydroxyurea, and inhibition of the ATR kinase. Furthermore, NEK8 was required for proper replication fork protection following replication stall with hydroxyurea. Loading of RAD51 to chromatin was decreased in NEK8-depleted cells and Nek8-knockout cells. Single-molecule DNA fiber analyses revealed that nascent DNA tracts were degraded in the absence of NEK8 following treatment with hydroxyurea. Consistent with this, Nek8-knockout cells showed increased chromosome breaks following treatment with hydroxyurea. Thus, NEK8 plays a critical role in replication fork stability through its regulation of the DNA repair and replication fork protection protein RAD51.
Introduction
Defects in DNA repair play a critical role in development of cancer and sensitivity of cancer cells to anti-cancer drugs. Homologous recombination (HR) is a mechanism of DNA repair that utilizes the undamaged homologous sequence as a template to repair double strand breaks (DSBs).Citation1 HR is particularly important for preventing cancer development, as exemplified by the observation that the genes regulating HR, such as BRCA1 and BRCA2, are tumor suppressor genes linked to breast/ovarian cancer.Citation2,3 Cancer cells that are defective in HR are sensitive to various anti-cancer drugs including interstrand DNA crosslinking agents (cisplatin, carboplatin and mitomycin C (MMC)) and poly (ADP-ribose) polymerase (PARP) inhibitors.Citation4 Therefore, a better knowledge of HR regulation will further our understanding of both cancer development and cancer therapy.
In higher eukaryotes, the genetic inactivation of many HR genes leads to lethality during the very early stages of development, suggesting that these proteins likely play key roles in DNA replication or in the repair of replication errors.Citation5 HR is required for the restart of replication forks in fission yeast, but the specific role of HR in replication fork restart in higher eukaryotes remains elusive.Citation6,7 One key HR protein involved in DNA replication is the recombinase RAD51, which is also a Fanconi anemia protein, FANCR.Citation8 RAD51 localization to active replication forks is required to prevent the accumulation of single-stranded DNA (ssDNA) regions directly at the fork. It is also hypothesized that this accumulation of RAD51 is required to prevent MRE11-dependent degradation of nascent DNA, which allows for continuous DNA replication.Citation9 Other proteins involved in the Fanconi anemia and HR pathway also have important roles in replication fork protection. For example, BRCA2/FANCD1 is directly required for replication fork protectionCitation10 through its recruitment of RAD51 to protect nascent DNA.Citation11
NEK8 is a member of the human NIMA-related kinase (NEK) family,Citation12 which contains 11 serine/threonine protein kinases. NEKs have mainly been studied in relation to their role in cell cycle progression, centrosome regulation, and ciliogenesis. NEK8 contains an N-terminal kinase domain. NEK8 differs from all but one other NEK kinase, NEK9, in that its C-terminal domain contains motifs similar to the regulator of chromatin condensation protein, RCC1.Citation13 A missense mutation in the RCC1 domain of Nek8 is reported as phenotypically causative in the mouse model of juvenile cystic kidney disease (JCK), where ciliary localization of the mutant Nek8 protein is defective.Citation14 Similarly, mutations in the conserved RCC1 domain of Nek8 are causative of the phenotypes observed in the rat model of Lewis polycystic kidney disease.Citation15 Germline mutations have been identified in human NEK8 that are implicated in the childhood autosomal recessive kidney disease nephronophthisis (NPHP),Citation16 in 3 patients with Ivemark syndrome, which is similar to polycystic kidney disease,Citation17 and in patients initially believed to have Alagille syndrome.Citation18 Most recently, novel NEK8 mutations were identified in 5 familial ciliopathy cases, where NEK8 missense mutations cause increased γH2AX foci, suggesting defects in DNA repair, which may lead to increased apoptosis during cell proliferation.Citation19 Furthermore, a missense mutation of NEK8 is reported as a potential driver mutation in pancreatic cancerCitation20 and NEK8 is overexpressed in human breast cancer.Citation21 These findings suggest a role of NEK8 in cancer development.
Intriguingly, NEK8 localizes not only to the centrosomes Citation16 and primary cilium,Citation22 but also to the nucleus.Citation12 Nuclear functions of NEK8 had not been studied until recently where NEK8 was linked to the ATR-mediated replication stress response via regulation of the protein kinase CDK2.Citation23 Cells deficient in NEK8 are characterized by an increase in histone H2AX phosphorylation, a sign of spontaneous DSBs. These DSBs further accumulate when replication forks stall. NEK8-deficient cells also exhibit reduced replication fork rates, unscheduled origin firing, and increased replication fork collapse.Citation23 As a result, NEK8-deficient cells are sensitive to replication inhibition by aphidicolin, a phenotype which is rescued by inhibition of CDK activity. Interestingly, NEK8 interacts with the checkpoint kinase ATR, CHK1 and the ATR interacting partner, ATRIP.Citation23 Furthermore, kidneys of Nek8-mutant mice accumulate DNA damage, and loss of Nek8 or replication stress similarly disrupts renal cell architecture in these mice.Citation23 Therefore, there exists evidence that NEK8 functions in the DNA damage response and DNA replication.
Phosphorylation plays an important role in the regulation of the DNA damage and replication stress responses. Several kinases have been implicated in the regulation of HR,Citation24-27 and multiple high-throughput screens have been used to identify factors required for HR and the replication stress response.Citation28-31 However, a comprehensive analysis of kinases involved in the regulation of HR following damage with an inter-strand cross-linking agent has not been reported leaving our understanding of the regulation of HR by phosphorylation incomplete.
Here we show that NEK8 is a critical regulator of RAD51 focus formation following DNA damage and genome stability following replication stress.
Results
Kinome siRNA screening identified NEK8 as a protein important for DNA damage-induced RAD51 foci formation
To identify novel kinases that regulate DNA damage-induced RAD51 foci formation, initially we performed an siRNA kinome library screen. The siRNA library contained 3 siRNA duplexes per gene targeting 713 human kinases. We performed the screen using inhibition of MMC-induced RAD51 foci formation in the human osteosarcoma cell line, U-2 OS, as readout. RAD51 foci were quantified with an automated high-throughput fluorescence microscope equipped with foci counting software (). The average number of foci per cell was converted to normalized z-scores ( and Table S1), where we focused on kinases with negative z-scores. Negative z-scores are indicative of kinase depletion leading to inhibited RAD51 foci formation following DNA damage. As RAD51 foci form mainly in S and G2 phases of the cell cycle,Citation32 we evaluated the percentage of cells in S phase following kinase depletion, using cyclin A immunostaining (Fig. S1 and Table S1). A total of 20 kinases were selected as positive hits (inhibition of RAD51 foci without decrease of cyclin A positive cells) in the primary screen (Table S1). To validate the result, the pooled siRNAs were de-convoluted and the 3 independent siRNAs against each candidate kinase were used to confirm the result in multiple cell lines (Fig. S1C and S2A-B).
Figure 1. Kinome siRNA screening identified NEK8 as a protein important for DNA damage-induced RAD51 foci formation. A. Screening strategy for identification of kinases required for RAD51 focus formation. U-2 OS cells were transfected with a siRNA kinome library, treated with MMC, immunostained and imaged for RAD51 focus formation. Representative images of RAD51 focus formation via automated acquisition and quantitation are shown. B. The mean Z-score of each kinase from the RAD51 foci screen are ranked (n = 2). C. Representative image of MMC (60 ng/mL, 24h)-induced RAD51 foci in U-2 OS cells. D. Quantification of the average number of RAD51 foci observed per U-2 OS cell following treatment with MMC (60 ng/mL, 24h). E. U-2 OS cells were transfected with siRNA (20nM). RNA was collected, converted to cDNA and amplified by PCR to confirm depletion. (n = 3, +/- SEM). * = p < 0.05, ** = p < 0.01, *** = p < 0.001 relative to siControl.
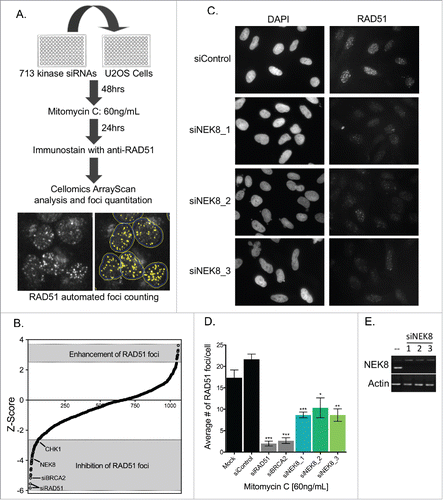
Among those genes whose siRNAs decreased MMC-induced RAD51 foci formation, NEK8, PAK4 and DGKD emerged as promising candidates ( and S1C). We chose to focus on NEK8 as other NIMA-related kinases had been implicated in proper cell cycle progression and the DNA damage response, Citation33 and NEK8 depletion by 3 independent siRNAs led to inhibition of MMC-induced RAD51 foci formation, while efficiently reducing NEK8 mRNA levels ().
NEK8 modulates RAD51 foci formation following DNA damage and replication fork stall
To determine if the effect NEK8 depletion had on RAD51 foci formation was specific to interstrand cross-links created by MMC, we next treated NEK8-depleted U-2 OS cells with various DNA damaging agents (Fig. S3A-D). RAD51 foci were also reduced in NEK8-depleted cells after treatment with ionizing radiation (IR) () as well as the replication inhibitor, hydroxyurea (HU) (), suggesting that the effect of NEK8 depletion on RAD51 foci formation is not limited to interstrand cross-links. We also tested Nek8−/− mouse embryonic fibroblasts (MEFs) and control MEFs for Rad51 foci formation following DNA damage. Reduced DNA damage-induced Rad51 foci formation was observed in Nek8−/− MEFs in all conditions tested (). Moreover, we observed similar phenotypes in the additional human cell line, HeLa, following treatment with MMC and HU (Fig. S4), suggesting that the effect of NEK8 deficiency on RAD51 foci formation is not cell type or species specific.
Figure 2. NEK8 modulates RAD51 foci formation following DNA damage and replication fork stall. A. Representative image of IR (10Gy, 6h)-induced RAD51 foci in U-2 OS cells treated with indicated siRNA. B. Quantification of the percentage of siRNA depleted U-2 OS cells with greater than 5 RAD51 foci following treatment with IR (10Gy, 6h), MMC (60 ng/mL, 24h) and HU (2 mM, 24 h). C. Western blot of key DNA repair protein expression in U-2 OS cells depleted of NEK8. D. siRNAs targeting NEK8 were transfected into U-2 OS cells (20nM), treated with HU (2 mM, 24 h) or untreated, then fixed and stained with propidium iodide prior to FACS cell cycle analysis. E. Representative image of IR (10Gy, 6 h) induced RAD51 foci in Nek8+/+ and Nek8−/− MEFs. F. Quantification of the percentage of Nek8+/+ and Nek8−/− MEFs with greater than 5 RAD51 foci following treatment with IR (10Gy, 6 h), MMC (60 ng/mL, 24 h) and HU (2 mM, 6 h). G. Western blot of key DNA repair protein expression in Nek8+/+ and Nek8−/− MEFs. H. Nek8+/+ and Nek8−/− MEFs were treated with HU (2 mM, 24 h) or untreated, harvested, fixed and stained with propidium iodide prior to FACS cell cycle analysis. (n = 3, +/- SEM). * = p < 0.05, ** = p < 0.01, *** = p < 0.001 relative to siControl or Nek8+/+ MEFs.
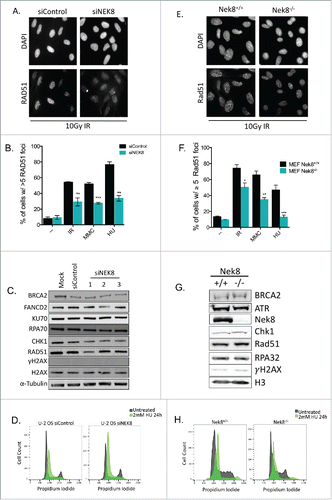
As formation of RAD51 foci is contingent on many key upstream factors and events in the HR pathway,Citation1 we analyzed the effect of NEK8 depletion on the expression of a few of these factors. Depletion of NEK8 did not affect the expression of the proteins important for RAD51 foci formation, such as BRCA2, RPA, and CHK1 (). Similar results were observed in Nek8−/− MEFs (). Furthermore, HU induced RPA foci form normally in NEK8-depleted U 2-OS cells (Fig. S5A) and in Nek8-knockout MEFs compare with control MEFs (Fig. S5B) following treatment with HU.
We noted a mild decrease in RAD51 protein expression following depletion of NEK8 with one of the NEK8 siRNAs (siNEK8_1) in U-2 OS cells (), but other two NEK8 siRNAs did not affect RAD51 protein expression level and Rad51 protein expression was not decreased in Nek8-knockout MEFs compare with control MEFs ().
Depletion or knockout of NEK8 did not significantly alter cell cycle profiles of U-2 OS cells or MEFs in untreated ( and H, gray profiles) and HU-treated ( and H, green profiles) conditions, signifying that the effect of NEK8 depletion on RAD51 foci formation is not mediated by cell cycle change. Together, these data demonstrate that NEK8 is a critical factor for RAD51 foci formation following DNA damage. The specificity of effect of NEK8 on RAD51 foci formation is not damage type, cell type, or species specific, but appears to be universal in nature.
NEK8 modulates resistance to DNA damaging agents and replication stress
As a key component of the HR pathway, tightly controlled regulation of RAD51 function mediates the resistance to various anti-cancer drugs including inter-strand DNA crosslinking agents and PARP inhibitors.Citation4 We tested the effect of loss of Nek8 in MEFs on resistance to multiple DNA damaging agents (). Nek8−/− MEFs were sensitive to the replication inhibitor, HU, the ATR kinase inhibitor, VE-821 and mildly to MMC and the PARP inhibitor, AZD2281 (). Consistent with this, U-2 OS cells depleted of NEK8 were mildly sensitive to HU (Fig. S6). Nek8−/− MEFs were not sensitive to a topoisomerase inhibitor, etoposide ().Citation34 Additionally, Nek8−/− MEFs were sensitive to the microtubule inhibitor, paclitaxel, suggesting a role for Nek8 during mitosis.
Figure 3. NEK8 is required for resistance to replication stress and acts independently of ATR. A. Cell survival in Nek8+/+ and Nek8−/− MEFs in response to increasing doses of hydroxyurea, MMC, PARP inhibitor (AZD2281), ATR inhibitor (VE-821), etoposide or paclitaxel as indicated. Cell survival was assayed by crystal violet staining and expressed as a fraction of the untreated control (n = 3, +/- SEM). B. U-2 OS cells were transfected with siRNA (20nM) and treated with or without HU (2 mM 24 h). Cells were then subjected to chromatin fractionation (S, soluble fraction; P, insoluble fraction (chromatin fraction)) and Western blotting. C. Nek8+/+ and Nek8−/− MEFs were treated with or without HU (2 mM 6 h) and then subjected to chromatin fractionation (S, soluble fraction; P, insoluble fraction (chromatin fraction)) and Western blotting. D. Cell survival in Nek8+/+ and Nek8−/− MEFs in response to a 6 h pulse of HU, with or without ATR inhibition followed by release into fresh media. Cell survival was assayed by crystal violet staining and expressed as a fraction of the untreated control (n = 7, +/-SEM).
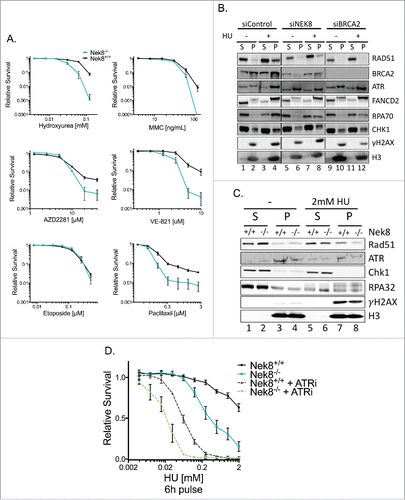
As NEK8-deficient cells were sensitive to HU, in line with a previous report showing NEK8 sensitivity to the replication inhibitor aphidicolin,Citation23 we hypothesized that NEK8 regulates the function of RAD51 in replication fork protection. To test this hypothesis, we treated NEK8-depleted U-2 OS and control cells with HU and performed cellular fractionation and western blotting to determine the efficiency of chromatin loading of RAD51. As expected, following treatment with HU, RAD51 chromatin loading was observed in control cells, while RAD51 chromatin loading was inefficient in NEK8-depleted U-2 OS cells ( lanes 4 and 8). Similarly, chromatin loading of Rad51 was inefficient in Nek8−/− MEFs compare with control MEFs (, lanes 7 and 8). The effect on chromatin loading of RAD51 was not limited to treatment with HU as we observed similar effects on loading of RAD51 in U-2 OS cells following treatment with MMC (Fig. S7, lanes 4 and 12). Additionally, we observed a decrease in chromatin loading of BRCA2 in NEK8-depleted U-2 OS cells (, lanes 4 and 8; Fig. S7, lanes 4 and 12) suggesting that the role of NEK8 in RAD51 chromatin loading may be due to a direct effect on BRCA2. However, this phenotype is not fully reproducible in the Nek8−/− MEF background (data not shown), therefore, we are hesitant to make this conclusion with no further evidence to support this hypothesis.
RAD51 has a non-canonical function in the early stages of replication fork protection, where it is loaded onto a reversed replication fork.Citation9,10,35-37 To test if NEK8 modulates this function of RAD51 at stalled replication forks, we treated Nek8−/− MEFs with HU for 6 hours, a time point at which DSB formation is minimal, Citation37 and then released the cells into fresh media. We then allowed the cells to recover and measured their relative survival. Nek8−/− MEFs were hypersensitive to HU under these conditions compared with Nek8+/+ MEFs (), suggesting that Nek8 has a function in the early response to replication fork stall.
Similar to NEK8 deficiency leading to hypersensitivity to HU, recent data shows that acute inhibition of ATR causes rapid cell death in cells experiencing replication stress.Citation38 As NEK8 has been linked to the ATR-regulated replication stress response via its regulation of CDK activity during S-phase of the cell cycle, Citation23 and our observation that Nek8−/− MEFs are sensitive to ATR inhibition (), we set out to further explore the role NEK8 has in the ATR-mediated replication stress response. We first verified that Nek8 is not required for proper checkpoint activation via phosphorylation on S345 of CHK1 (Fig. S8A) as reported by Liu et. al.Citation39 We next validated that Nek8 is required for resistance to ATR inhibition by rescuing this phenotype through exogenous expression of wild-type Nek8 in Nek8−/− MEFs (Fig. S8B-C). Lastly, we tested the effect of ATR inhibition on cellular survival following treatment with HU in Nek8−/− MEFs. ATR inhibition further sensitized Nek8−/− MEFs to HU (), suggesting that NEK8 and ATR are independently important for cellular survival after replication inhibition.
Taken together, these data suggest that NEK8 plays an important role in cellular resistance to replication stress, in the recruitment of the essential replication fork protection factor RAD51 to chromatin and acts independently of ATR. This role of NEK8 may be an essential early response to replication fork stall mediated by RAD51.
NEK8 preserves genome stability
Cells having defective Fanconi anemia and HR pathways also exhibit increased levels of replication fork degradation following treatment with replication fork inhibitors and increased levels of genomic instability in response to replication stress.Citation10,11 NEK8 also regulates replication fork speed, stability and origin firing.Citation23 However, the mechanism by which this process is regulated is not fully understood. On this basis, we hypothesized that NEK8 is maintaining replication fork protection and genomic stability via its role in the proper function of RAD51 at stalled replication forks.
To test this hypothesis, we first used DNA fiber analysis to investigate replication fork dynamics in Nek8−/− MEFs. Using a dual labeling technique followed by treatment with HU, we measured both normal progression of replication (IdU tract length) and replication fork stability via degradation (CldU tract length) (). In our hands, Nek8−/− MEFs did not exhibit a significant difference in replication tract length compare with Nek8+/+ MEFs as measured by IdU incorporation (). This is in contrast to a previous report showing Nek8−/− MEFs having slowed rates of fork progression.Citation23 Our results may differ due to the different types of assays used.
Figure 4. Nek8 prevents replication fork degradation following replication fork stall. A. Schematic of experimental conditions for DNA replication tract assay. Red tracts, IdU; Green tracts, CldU. B. IdU tract length in HU treated or untreated Nek8+/+ and Nek8−/− MEFs. Median tract length is designated in box and whisker plots. C. CldU tract length in HU treated or untreated Nek8+/+ and Nek8−/− MEFs. Mean tract length is denoted in parenthesis and median tract length is designated in box and whisker plots. D. Schematic of experimental conditions for DNA replication tract assay treated with Mirin. Green tracts, CldU. E. CldU tract length in HU treated (4mM, 5 h) and Mirin treated or untreated Nek8+/+ and Nek8−/− MEFs. Mean tract length is denoted in parenthesis and median tract length is designated in box and whisker plots F. Representative DNA fiber images from Nek8+/+ and Nek8−/− MEFs either treated or untreated with HU. (n=∼300 fibers/sample) *** = p < .001, * = p < .05.
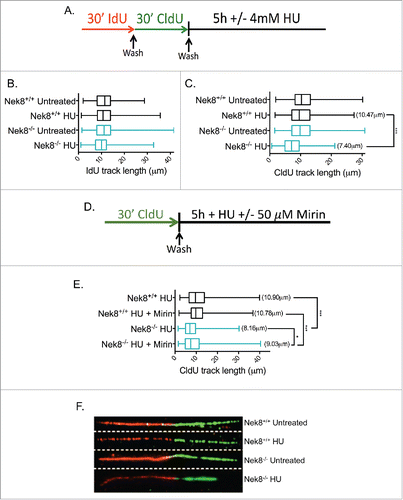
We next measured the length of CldU tracts following treatment with HU. Nek8−/− MEFs exhibited significantly shorter CldU tract lengths following HU as compare with Nek8+/+ MEFs (). This suggests that resection of DNA at stalled replication forks is increased in the absence of Nek8. The nuclease MRE11 has been implicated in the degradation of stalled replication forks in the absence of proper fork protection by members of the Fanconi anemia and HR pathways.Citation10,11 To determine if replication tract degradation in Nek8−/− MEFs is also mediated by MRE11, we measured degradation (CldU tract length) in cells treated with HU and with or without the MRE11 inhibitor, Mirin (). Nek8−/− MEFs treated with HU exhibit shorter CldU tract lengths as compared with Nek8+/+ MEFs. This phenotype is incompletely rescued following treatment with Mirin (), suggesting that MRE11 may play a role in mediating resection of stalled replication forks in Nek8−/− MEFs, but is not the only factor involved.
Next, the observation that spontaneous DNA damage accumulates in NEK8-deficient cells,Citation23 coupled with the assumption that HR is required for the repair of DNA replication-associated spontaneous DNA damage,4041 led us to test the efficiency of HR in the absence of NEK8. Using the U-2 OS DR-GFP reporter assay,Citation42 we found that depletion of NEK8 via siRNA led to a roughly 2-fold decrease in efficient HR when compare with control siRNA-transfected U-2 OS cells ().
Figure 5. Nek8 is important for homologous recombination efficiency and the maintenance of genome stability. A. U-2 OS DR-GFP cells were transfected with siRNA (20nM). 24 h post transfection cells were transfected with pCBASce or control plasmids. GFP expression was detected and quantified via flow cytometry. Representative GFP+ population is denoted as a percentage. B. Quantification of U-2 OS DR-GFP assay using relative GFP expression (n = 3, +/- SEM, *p<.05, **p<.01). C. Representative images of metaphase chromosomes of Nek8+/+ and Nek8−/− MEFs treated with or without HU for 6 h and then released into fresh media for 16 h. The cells were then treated with colcemid and processed for metaphase analysis. D. Quantification of mean chromosome aberrations per metaphase (aberrations include: breaks, gaps, radials and other translocations) (n = 30 metaphases per sample, +/- SEM).
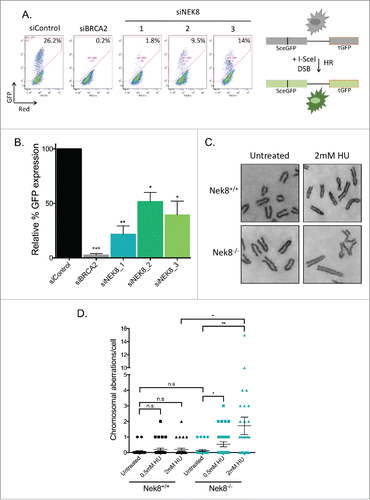
Defects in replication fork protection mediated by proteins of the Fanconi anemia and HR pathways increases genomic instability in response to HU.Citation10,11 Therefore, we next tested if treatment with HU increased levels of genomic instability in Nek8−/− MEFs via metaphase spread analysis. The difference of basal levels of chromosomal aberrations was not statistically significant between Nek8+/+ and Nek8−/− MEFs in the absence of replication inhibition by HU (, 0.06 ± 0.04 and 0.13 ± 0.06, p = 0.398). However, after treatment with increasing doses of HU, Nek8−/− MEFs showed a drastic increase in the number of chromosomal aberrations per cell compared with Nek8+/+ MEFs (, 0.2 ± 0.08 and 1.71 ± 0.55, p = 0.011).
Taken together, these data suggest that NEK8 is a critical factor for the maintenance of genomic stability mediated through HR and replication fork protection.
Discussion
In this study, we conducted a high-throughput siRNA library screen of the human kinome using RAD51 focus formation as a readout and identified the NIMA-related kinase, NEK8, as a regulator of RAD51 foci formation and replication fork stability. NEK8-deficient cells had decreased RAD51 foci formation in response to MMC, IR and HU and showed decreased HR efficiency and an increase in chromosomal aberrations. We found that cells deficient in NEK8 were sensitive to the replication stress-inducing agent HU (), consistent with previous reports showing that NEK8 deficiency renders cells sensitive to aphidicolin.Citation23
Among the human NEK family kinases, NEK8 along with other family members, NEK1, NEK6, and NEK11, have been implicated in the DNA damage response, though to differing degrees.Citation23,39-43,49 NEK6 inhibition is required for proper activation of cell cycle arrest following DNA damage,Citation47 while activation of NEK1 and NEK11 is required for DNA damage checkpoint control, DNA damage repair and the response to damage induced by ultraviolet radiation, IR, cisplatin and other drugs.Citation44,46,50,51 NEK1 also orchestrates HR and replication fork stability via regulation of RAD54.Citation43 Similarly, the NIMA-related kinase in Saccharomyces cerevisiae, Kin3, is involved in the response to DNA adduct damage.Citation52 Our findings suggest that NEK8 further links the NIMA-related kinase family to the DNA damage response, where NEK8 maintains a complex regulatory role in the repair of DNA lesions requiring HR as well as having a significant role in the protection of replication forks.
Interestingly, we observed hypersensitivity to short-term replication stress in Nek8−/− MEFs (). This phenotype suggests that Nek8 has an important function in the early stages of replication fork protection, possibly mediated through RAD51. Previous data suggests that key components of the Fanconi anemia and HR pathways, including RAD51, have important roles in the early stages of replication fork protection and recovery, possibly mediated by the HR-associated protein, RAD18.Citation10,11,37,53 The cell cycle checkpoint kinase, ATR, is also an important factor in replication fork protection.Citation54-57 NEK8 interacts with ATR, ATR-interacting protein (ATRIP), and CHK1.Citation23 Previous data also suggests that proper regulation of RAD51 expression modulates DNA replication and replication fork protection in a CHK1-dependent manner.Citation58 We observed that Nek8−/− MEFs were sensitive to inhibition of ATR and were further sensitized to HU following ATR inhibition (). This suggests that NEK8 has a separate and distinct function from ATR in the early stages of replication fork protection and possibly in the maintenance of chromosomal stability. Precise molecular mechanisms by which NEK8 cooperates with these factors (ATR, CHK1, RAD18, RAD51 and other Fanconi anemia proteins) in the replication fork protection remain to be elucidated.
We observed an increase in degradation of stalled replication forks in the Nek8−/− MEFs (). This phenotype is not completely unexpected as previous reports show that protection of nascent DNA at stalled replication forks requires key factors in the Fanconi anemia and HR repair pathways.Citation10,11 We found that the nuclease MRE11 may play a role in degradation of stalled replication forks in the absence of Nek8 (). However, MRE11 is likely not the only factor responsible, since the MRE11 inhibitor, mirin, only partially restored fork protection in HU-treated Nek8−/− MEFs. Other nucleases, including DNA2 and MUS81, as well as other factors required for replication fork protection, such as BODL1,Citation57 could also be involved in regulating replication fork degradation in Nek8−/− MEFs. Nek8−/− MEFs also exhibited an increase in genomic instability following treatment with HU (). This phenotype is likely explained by a decrease in HR () coupled with the inability to efficiently restart replication after replication inhibition,Citation23 ultimately manifesting as genome instability and cellular sensitivity to replication inhibiting drugs. It would be worthwhile to explore the functional domains of NEK8 which are required for the observed phenotypes in our experimental system. Previous reports suggest the importance of both the kinase domain and RCC1 domains of NEK8 as being important for its localization to the ciliary axoneme, and as such, its function in NPHP, as well as for its role in the ATR-mediated replication stress response.Citation12,23 Understanding the functional significance of these domains in replication fork protection and genome stability would provide mechanistic insight and a foundation for the future development of therapeutic strategies targeting NEK8.
Lastly, NEK8 localizes to the centrosomes,Citation16 primary cilium Citation22 and nucleus.Citation12 Interestingly, the functional interaction between DNA repair proteins and centrosome proteins is an emerging concept.Citation59 Several DNA repair proteins including BRCA1, BRCA2 and RAD51, localize to the centrosome, in addition to the nucleus, and are involved in the regulation of multiple processes, such as centrosome functionality.Citation59,60 Mutations in genes which cause ciliopathies, including the centrosomal protein CEP164,Citation61,62 ZNF423, MRE11 and FAN1Citation61-63 also have important functions in the DNA damage response, suggesting a link between centrosomes, ciliopathies and DNA repair pathways. The observation that Nek8−/− MEFs were sensitive to the microtubule poison drug paclitaxel (), suggests that similar to the other NEK family kinases, NEK2, NEK5 and NEK6,Citation64 NEK8 may have a functional role in proper mitotic chromosome segregation, whose errors may drive cancer progression. This observation also further links the DNA damage response, the replication stress response and ciliopathies.
In summary, we describe multiple phenotypes associated with the loss of NEK8 in mammalian cells. Our data highlight important functions of NEK8 in the replication stress response. The identification of NEK8 as a regulator of HR and replication stress response provides further insight into the intricate regulatory mechanisms behind an already complex cellular machine responsible for the response to DNA damage and replication stress. However, these data also raise questions about the role of NEK8 in fine tuning the response to DNA damage, an area which remains cloudy. Furthermore, these findings provide further evidence to the interplay between centrosomal proteins and the DNA repair machinery, highlighting the importance for future research in this area.
Materials and methods
Cell lines
U-2 OS and HeLa were purchased from the American Type Culture Collections. Nek8−/− and Nek8+/+ MEFsCitation23,65 were a gift of the Cimprich lab (Stanford) with permission from the Beier lab (Seattle Children's). U-2 OS DR-GFP cells were a gift of the Jasin Lab (Memorial Sloan Kettering).Citation66 Cell lines were cultured in DMEM containing 10% FBS, 2 mM L-glutamine and 1X Pen/Strep in a humidified 5% CO2 containing atmosphere at 37°C.
siRNA kinome library screening
U-2 OS cells were transfected (20nM, Lipofectamine RNAiMAX) with the MISSION siRNA human kinase panel library (Sigma) in 384 well glass bottom cell culture plates (Thermo Scientific, #4331). Control siRNAs were spiked-in and included BRCA2,Citation67 RAD51 (5′-AACTAATCAGGTGGTAGCTCA-3′), ATR (5′-AACCTCCGTGATGTTGCTTGA-3′), and Chk1 (5′ AAGGGATAACCTCAAAATCTC-3′). Two days post-transfection, cells were treated with MMC (60 ng/mL) for 24 h. Cells were then simultaneously fixed and permeabilized (2% PFA and 0.5% Triton X-100 in PBS for 20 minutes) followed by immunostaining for RAD51 or cyclin A. Images were acquired with the Cellomics Arrayscan microscope (Thermo Scientific) and processed as described.Citation68 Using DAPI to define nuclei, the average number of foci per cell (RAD51) and nuclear intensity (cyclin A) was quantitated using the automated counting software. The Z-score was calculated based on the formula Z=(X-μnc)/σ, where X was the individual sample average, μnc was the mean of the negative control population in each plate and σ was the standard deviation of the whole population. Screening was independently replicated 2 times.
Immunofluorescence microscopy
Immunofluorescence microscopy was conducted as previously described.Citation68 Briefly, transfected cells were grown on coverslips, treated with MMC (60 ng/mL, 24 h), IR (10 gy, 6 h) or HU (2 mM, 24 h) and then simultaneously fixed and permeabilized (2% PFA and 0.5% Triton X-100 in PBS for 20 minutes). Cells were immunostained for RAD51 and RPA. Images were acquired with an inverted fluorescent microscope (TE2000, Nikon) and analyzed using ImageJ (National Institute of Health). At least 300 cells per experimental point were scored for the presence of foci. Each experiment was repeated 3 times independently.
RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) and reverse-transcribed using the SuperScript III First-Strand Synthesis system (ThermoFisher). cDNA of target was amplified via PCR with the primers: TEX14 (For 5′-TAGACTCCCCGCAGCGGCTT-3′, Rev 5′-TGAAACCAAGGCATAGCCTTCCC-3′), NEK8 (For 5′-GCAAGCCCTACAACCAGAAG-3′, Rev 5′-ACTCATGATCTTCAGCACCAG-3′), PAK4 (For 5′-CTCCTCGTTCATCCTGGTGT-3′, Rev 5′-GAGCTGCTCTTCAACGAGGT-3′), DGKD (For 5′-GCTTGTGCAAGAAGGAGGAC-3′, Rev 5′-ACTGTGTGCGACAAGACCTG-3′), RPS6KL1 (For 5′-AAGGGGTCACTGTGAGGATG-3′, Rev 5′-AGGCTCCCCTGTAGAAGCTC-3′), FUK (For 5′-CTGGAGCCATGAGCTTCTTC-3′, Rev 5′-TGTGCTGAGGAGCTGGTATG-3′), PTK6 (For 5′-CTGCAGACAGACAGCCAGAG-3′, Rev 5′-CCTGGGGTTTACTGAGGTGA-3′). Amplified targets were resolved on a 2% agarose gel. Images of gels were obtained with a UV imager (BioRad Gel Doc XR+).
Western blotting
Whole-cell extracts were prepared and resolved by polyacrylamide gel electrophoresis as described.Citation68 Proteins were transferred onto nitrocellulose membranes. Antibodies against α-Tubulin (CST), Actin (sc-1616-R, Santa Cruz), ATR (N-19, Santa Cruz), BRCA1 (D-9, Santa Cruz), BRCA2 (Ab-2, Calbiochem), CHK1 (G-4, Santa Cruz), FANCD2 (Abcam), FLAG (M-2, Santa Cruz), γH2AX (JBW301, Millipore), GFP (Life Technologies), H2AX (Millipore), H3 (Abcam), Ku70 (Abcam), mNEK8 (gift from David Beier),Citation14 hNEK8 (N-17, Santa Cruz), RAD51 (H-92, Santa Cruz and BAM-10–001, CosmoBio) and RPA70 (CST) were probed with horseradish peroxidase-conjugated anti-mouse, anti-rabbit (GE Biosciences) or anti-goat IgG (sc-2020, Santa Cruz). Chemiluminescence was used for detection and membranes were digitally scanned with an Imagequant LAS 4000 (GE Biosciences). Images were processed using Photoshop CS (Adobe Systems, Inc.) and PowerPoint (Microsoft, Inc.).
Cell cycle analysis
siRNA-transfected U-2 OS cells or Nek8-knockout MEFs were treated with HU (2 mM,24 h) or not treated. Cells were then fixed and stained with propidium iodide for DNA content. Flow cytometry was performed to determine the cell cycle phase distribution (Canto 2 or LSR-2) and cell cycle profiles were obtains with FloJo software.
siRNAs and plasmids
siRNAs targeting BRCA2,Citation67 RAD51 (target sequence: 5′- AACTAATCAGGTGGTAGCTCA-3′), and NEK8 (#1 target sequence: 5′- TCACTCTTCTGGTTGTAGG-3′, #2 target sequence: 5′- TCAGAGGAGAAGCAATATC-3′, #3 target sequence: 5′- AGAGATAGGTGCAAAGGTG-3′) were transfected at 20nM using Lipofectamine RNAiMAX (ThermoFisher). AllStars siRNA (Qiagen) was used as a negative control. I-SceI expression vector, pCBASce, was a gift from the Jasin lab (Memorial Sloan Kettering). mNek8 plasmid was a gift from the Beier lab (Seattle Children's). mNek8 coding sequence was PCR amplified from the mNek8 plasmid and cloned into pMMP-IRES-puro. Retroviruses were produced as previously described.Citation69 Nek8+/+ and Nek8−/− MEFs were transduced with pMMP-IRES-puro (empty vector) or pMMP-IRES-puro-mNek8 retroviruses were selected with puromycin at 3 μg/mL for 3 d.
Homologous recombination assay
U2OS DR-GFP cells were transfected with siRNAs and then transfected 24 hours later with pCBASce (I-SceI expression vector). Two days after plasmid transfection, cells were harvested and analyzed using a FACSCalibur analyzer or an LSRII analyzer to determine the percentages of GFP- positive cells.
Cell fractionation
Cell fractionations were prepared as described.Citation70 Briefly, cells were resuspended in buffer CSK (10mM PIPES, pH = 6.8, 100mM NaCl, 1mM EGTA, 1mM EDTA, 300mM Sucrose, 1.5mM MgCl2, 0.1% Triton-X-100 and protease inhibitors) and incubated in ice for 5 min. Samples were centrifuged at 1500 g for 5 min. Supernatant was collected and stored (soluble fraction). Pellets (pellet fraction) were washed once in CSK buffer and then re-suspended in sample buffer (0.05 M Tris-HCl (pH 6.8), 2% SDS, 6% β-mercaptoethanol) and boiled for 5 min.
Survival assay
Cell survival was measured by a crystal violet absorbance-based assay. Cells were seeded onto 12-well plates at a density of 6–9×106 cells/well. The next day, cells were treated with increasing concentrations of drug and incubated for 5 to 8 more days. After that, cellular monolayers were fixed, stained with crystal violet and re-solubilized as previously described.Citation68
DNA fiber assay
DNA fiber assay was conducted as previously described with some changes.Citation10 Briefly, cells were labeled with IdU (50 μM), washed, and labeled with CldU (50 μM). Cells were then exposed to HU (4 mM), Mirin (50 μM) or untreated media. DNA fibers were essentially spread as describedCitation71 before standard detection of IdU and CldU tracts (primary antibodies: α-IdU, α-BrdU from BD Biosciences; α-CldU, α-BrdU from Novus Biologicals and secondary antibodies: Alexa Fluors 488 and 594, respectively, from Invitrogen). Fibers were imaged on an inverted fluorescent microscope (TE2000, Nikon) and analyzed using ImageJ software. Statistics were calculated using Prism6 (GraphPad Inc.).
Metaphase spread analysis
Seven × 105 cells were seeded 24 h prior to treatment with HU (4 mM) and treated with colcemid (0.1μg/ml, GIBCO). Cells were swollen with 0.075M KCL (15min, 37°C), fixed with methanol/acetic acid (3:1), dropped onto a microscope slide, stained with 5% Giemsa, and mounted with ENTELLAN NEW (Electron Microscopy Sciences) before imaging with an inverted fluorescent microscope (TE2000, Nikon).
Statistical analysis
A Student's t-test was used to evaluate significance of differences in all experiments (Excel, Microsoft, Inc. and Prism6, GraphPad, Inc.). All experiments were expressed as mean ± SEM. A P value < 0.05 was considered significant.
Abbreviations
| DSB | = | double strand break |
| HR | = | homologous recombination |
| HU | = | hydroxyurea |
| IR | = | ionizing radiation |
| JCK | = | juvenile cystic kidney disease |
| MEFs | = | mouse embryonic fibroblasts |
| MMC | = | mitomycin C |
| NEK | = | NIMA-related kinase |
| NPHP | = | nephronophthisis |
| PARP | = | poly (ADP-ribose) polymerase |
| ssDNA | = | single-stranded DNA |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCCY_A_1259038_Supplementary_material.zip
Download Zip (5.6 MB)Acknowledgments
We thank Drs. Maria Jasin, David Beier and Karlene Cimprich for reagents, James Annis and Dr. Julia Sidorova for technical assistance, and Drs. Oliver Fregoso and Nick Wallace for helpful discussions. We thank all members of the Taniguchi laboratory for technical support and helpful discussions.
Funding
This work was supported by Howard Hughes Medical Institute, and the NIH/NCI [R01 CA125636 to T.T.]. A.A was supported by the National Science Foundation Graduate Research Fellowship under Grant No. [DGE-0718124] and the Fred Hutch CMCTG [T32 CA009657].
Related Research Data
References
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415
- Smith SA, Easton DF, Evans DG, Ponder BA. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat Genet 1992; 2:128-31; PMID:1303261
- Collins N, McManus R, Wooster R, Mangion J, Seal S, Lakhani SR, Ormiston W, Daly PA, Ford D, Easton DF, et al. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene 1995; 10:1673-5; PMID:7731724
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 2008; 26:3785-90; PMID:18591545
- Aze A, Zhou JC, Costa A, Costanzo V. DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 2013; 122:401-13; PMID:23584157
- Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Freon K, Murray JM, Carr AM, Baldacci G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 2010; 39:346-59; PMID:20705238
- Mizuno K, Lambert S, Baldacci G, Murray JM, Carr AM. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev 2009; 23:2876-86; PMID:20008937
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell 2015; 59:478-90; PMID:26253028
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 2010; 17:1305-11; PMID:20935632
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542
- Zalli D, Bayliss R, Fry AM. The Nek8 protein kinase, mutated in the human cystic kidney disease nephronophthisis, is both activated and degraded during ciliogenesis. Hum Mol Genet 2012; 21:1155-71; PMID:22106379
- O'Connell MJ, Krien MJ, Hunter T. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol 2003; 13:221-8.
- Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, Drummond IA, Beier DR. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002; 129:5839-46; PMID:12421721
- McCooke JK, Appels R, Barrero RA, Ding A, Ozimek-Kulik JE, Bellgard MI, Morahan G, Phillips JK. A novel mutation causing nephronophthisis in the Lewis polycystic kidney rat localises to a conserved RCC1 domain in Nek8. BMC Genomics 2012; 13:393; PMID:22899815
- Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol 2008; 19:587-92; PMID:18199800
- Frank V, Habbig S, Bartram MP, Eisenberger T, Veenstra-Knol HE, Decker C, Boorsma RA, Gobel H, Nurnberg G, Griessmann A, et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum Mol Genet 2013; 22:2177-85; PMID:23418306
- Rajagopalan R, Grochowski CM, Gilbert MA, Falsey AM, Coleman K, Romero R, Loomes KM, Piccoli DA, Devoto M, Spinner NB. Compound heterozygous mutations in NEK8 in siblings with end-stage renal disease with hepatic and cardiac anomalies. Am J Med Genet A 2015.
- Grampa V, Delous M, Zaidan M, Odye G, Thomas S, Elkhartoufi N, Filhol E, Niel O, Silbermann F, Lebreton C, et al. Novel NEK8 Mutations Cause Severe Syndromic Renal Cystic Dysplasia through YAP Dysregulation. PLoS Genet 2016; 12:e1005894; PMID:26967905
- Carter H, Samayoa J, Hruban RH, Karchin R. Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer Biol Ther 2010; 10:582-7; PMID:20581473
- Bowers AJ, Boylan JF. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 2004; 328:135-42; PMID:15019993
- Mahjoub MR, Trapp ML, Quarmby LM. NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J Am Soc Nephrol 2005; 16:3485-9; PMID:16267153
- Choi HJ, Lin JR, Vannier JB, Slaats GG, Kile AC, Paulsen RD, Manning DK, Beier DR, Giles RH, Boulton SJ, et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell 2013; 51:423-39; PMID:23973373
- Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP. Regulation of Rad51 function by phosphorylation. EMBO Rep 2011; 12:833-9; PMID:21738226
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005; 7:195-201; PMID:15665856
- Yata K, Esashi F. Dual role of CDKs in DNA repair: to be, or not to be. DNA Repair (Amst) 2009; 8:6-18; PMID:18832049
- Yata K, Lloyd J, Maslen S, Bleuyard JY, Skehel M, Smerdon SJ, Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol Cell 2012; 45:371-83; PMID:22325354
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol 2012; 14:318-28; PMID:22344029
- Kondo S, Perrimon N. A genome-wide RNAi screen identifies core components of the G(2)-M DNA damage checkpoint. Sci Signal 2011; 4:rs1; PMID:21205937
- Patrick Herr CL, Bastiaan Evers, Daniel Ebner, Christina Bauerschmidt, Guy Kingham, Timea Palmai-Pallag, Oliver Mortusewicz, Oliver Frings, Erik Sonnhammer & Thomas Helleday. A genome-wide IR-induced RAD51 foci RNAi screen identifies CDC73 involved in chromatin remodeling for DNA repair. Cell Discovery 2015; 1; PMID:27462432
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009; 35:228-39; PMID:19647519
- Tashiro S, Kotomura N, Shinohara A, Tanaka K, Ueda K, Kamada N. S phase specific formation of the human Rad51 protein nuclear foci in lymphocytes. Oncogene 1996; 12:2165-70; PMID:8668342
- Fry AM, O'Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci 2012; 125:4423-33; PMID:23132929
- Chernikova SB, Game JC, Brown JM. Inhibiting homologous recombination for cancer therapy. Cancer Biol Ther 2012; 13:61-8; PMID:22336907
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 2015; 208:563-79; PMID:25733714
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 2011; 25:1320-7; PMID:21685366
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 2013; 27:1610-23; PMID:23873943
- Liu S, Ho CK, Ouyang J, Zou L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A 2013; 110:2175-80; PMID:23345434
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998; 17:598-608; PMID:9430650
- Venkitaraman AR. Chromosome stability, DNA recombination and the BRCA2 tumour suppressor. Curr Opin Cell Biol 2001; 13:338-43; PMID:11343905
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13:2633-8; PMID:10541549
- Spies J, Waizenegger A, Barton O, Surder M, Wright WD, Heyer WD, Lobrich M. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol Cell 2016; 62:903-17; PMID:27264870
- Sorensen CS, Melixetian M, Klein DK, Helin K. NEK11: linking CHK1 and CDC25A in DNA damage checkpoint signaling. Cell Cycle 2010; 9:450-5; PMID:20090422
- Noguchi K, Fukazawa H, Murakami Y, Uehara Y. Nucleolar Nek11 is a novel target of Nek2A in G1/S-arrested cells. J Biol Chem 2004; 279:32716-27; PMID:15161910
- Noguchi K, Fukazawa H, Murakami Y, Uehara Y. Nek11, a new member of the NIMA family of kinases, involved in DNA replication and genotoxic stress responses. J Biol Chem 2002; 277:39655-65; PMID:12154088
- Lee MY, Kim HJ, Kim MA, Jee HJ, Kim AJ, Bae YS, Park JI, Chung JH, Yun J. Nek6 is involved in G2/M phase cell cycle arrest through DNA damage-induced phosphorylation. Cell Cycle 2008; 7:2705-9; PMID:18728393
- Chen Y, Chen CF, Riley DJ, Chen PL. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011; 10:655-63; PMID:21301226
- Chen Y, Chen CF, Chiang HC, Pena M, Polci R, Wei RL, Edwards RA, Hansel DE, Chen PL, Riley DJ. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Mol Cancer 2011; 10:5; PMID:21214959
- Polci R, Peng A, Chen PL, Riley DJ, Chen Y. NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res 2004; 64:8800-3; PMID:15604234
- Chen Y, Chen PL, Chen CF, Jiang X, Riley DJ. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008; 7:3194-201; PMID:18843199
- Moura DJ, Castilhos B, Immich BF, Canedo AD, Henriques JA, Lenz G, Saffi J. Kin3 protein, a NIMA-related kinase of Saccharomyces cerevisiae, is involved in DNA adduct damage response. Cell Cycle 2010; 9:2220-9; PMID:21577056
- Tripathi K, Mani C, Clark DW, Palle K. Rad18 is required for functional interactions between FANCD2, BRCA2, and Rad51 to repair DNA topoisomerase 1-poisons induced lesions and promote fork recovery. Oncotarget 2016; 7:12537-53; PMID:26871286
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029
- Hyrien O. Mechanisms and consequences of replication fork arrest. Biochimie 2000; 82:5-17; PMID:10717381
- Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol 2012; 47:222-35; PMID:22324461
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol Cell 2015; 59:462-77; PMID:26166705
- Parplys AC, Seelbach JI, Becker S, Behr M, Wrona A, Jend C, Mansour WY, Joosse SA, Stuerzbecher HW, Pospiech H, et al. High levels of RAD51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired CHK1 activation. Cell Cycle 2015; 14:3190-202; PMID:26317153
- Shimada M, Komatsu K. Emerging connection between centrosome and DNA repair machinery. J Radiat Res 2009; 50:295-301; PMID:19542690
- Cappelli E, Townsend S, Griffin C, Thacker J. Homologous recombination proteins are associated with centrosomes and are required for mitotic stability. Exp Cell Res 2011; 317:1203-13; PMID:21276791
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150:533-48; PMID:22863007; http://dx.doi.org/10.1016/j.cell.2012.06.028
- Sivasubramaniam S, Sun X, Pan YR, Wang S, Lee EY. Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes Dev 2008; 22:587-600; PMID:18283122; http://dx.doi.org/10.1101/gad.1627708
- Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012; 44:910-5; PMID:22772369; http://dx.doi.org/10.1038/ng.2347
- Prosser SL, O'Regan L, Fry AM. Novel insights into the mechanisms of mitotic spindle assembly by NEK kinases. Mol Cell Oncol 2016; 3:e1062952; PMID:27314078; http://dx.doi.org/10.1080/23723556.2015.1062952
- Manning DK, Sergeev M, van Heesbeen RG, Wong MD, Oh JH, Liu Y, Henkelman RM, Drummond I, Shah JV, Beier DR. Loss of the ciliary kinase Nek8 causes left-right asymmetry defects. J Am Soc Nephrol 2013; 24:100-12; PMID:23274954; http://dx.doi.org/10.1681/ASN.2012050490
- Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol 2006; 409:524-40; PMID:16793422; http://dx.doi.org/10.1016/S0076-6879(05)09031-2
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008; 451:1116-20; PMID:18264087; http://dx.doi.org/10.1038/nature06633
- Wang Y, Huang JW, Li M, Cavenee WK, Mitchell PS, Zhou X, Tewari M, Furnari FB, Taniguchi T. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Mol Cancer Res 2011; 9:1100-11; PMID:21693595; http://dx.doi.org/10.1158/1541-7786.MCR-11-0007
- Huang JW, Wang Y, Dhillon KK, Calses P, Villegas E, Mitchell PS, Tewari M, Kemp CJ, Taniguchi T. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol Cancer Res 2013; 11:1564-73; PMID:24088786; http://dx.doi.org/10.1158/1541-7786.MCR-13-0292
- Castella M, Jacquemont C, Thompson EL, Yeo JE, Cheung RS, Huang JW, Sobeck A, Hendrickson EA, Taniguchi T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet 2015; 11:e1005563; PMID:26430909; http://dx.doi.org/10.1371/journal.pgen.1005563
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 1998; 140:1285-95; PMID:9508763; http://dx.doi.org/10.1083/jcb.140.6.1285