
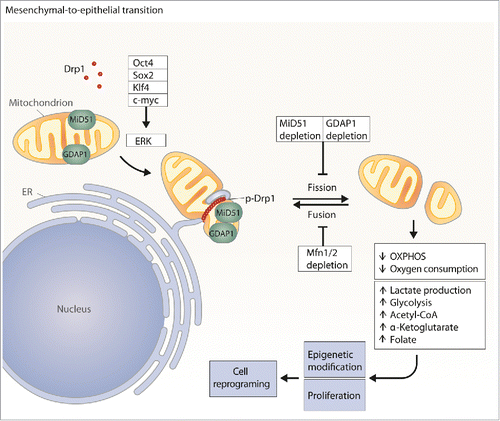
The OSKM (Oct3/4, Sox2, Klf4, and c-Myc) factors trigger a complex array of signals that promote the reprogramming of somatic cells. The modification of mitochondrial metabolism is a hallmark of cellular reprogramming. In this regard, during reprogramming, cells are characterized by high rates of glycolysis and lactate production, and elevated concentrations of acetyl-CoA, α-ketoglutarate and folate, together with reduced mitochondrial metabolism, respiration, and OXPHOS expressionCitation1,2 (). The metabolic modifications that occur during reprogramming are thought to sustain proliferation and also the epigenetic modifications required to maintain the induced pluripotent stem cell (iPSC) phenotype.
Figure 1. Mitochondrial fission during the early steps of cellular reprogramming. ERK is activated by OSKM factors, which induce Drp1 to promote mitochondrial fission at sites marked by the ER tubules. GDAP1 and MiD51 facilitate Drp1 function. Prieto et al., demonstrate that GDAP1 loss-of function prevents mitochondrial fragmentation and reduces cell reprogramming. Conversely, Mfn1 and Mfn2 promote mitochondrial fusion and sustain the somatic phenotype. Fragmented mitochondria are linked to reduced OXPHOS and promote the formation of key metabolites for cell reprogramming.
The plasticity of organelle architecture plays a key role in cellular metabolism. In this regard, mitochondrial dynamics—defined as the movement of mitochondria along the cytoskeleton, and the regulation of their architecture, and connectivity mediated by tethering and fusion/fission—sustains mitochondrial homeostasis. Several reports have demonstrated that the dysregulated expression of proteins involved in mitochondrial fission or fusion induce oxidative stress, mitochondrial dysfunction, and alterations in mitophagy. Additionally, mutations or reduced expression of proteins implicated in mitochondrial dynamics are associated with type 2 diabetes, Alzheimer disease, and cardiomyopathy. Mitochondrial dynamics also plays a role in cell proliferation, and during the G1/S phase, the mitochondrial network is elongated in order to support the bioenergetic requirements of DNA synthesis and of global cellular proliferation. Drp1, a GTPase that stimulates mitochondrial fission, promotes mitochondrial fragmentation during the M phase. This process is essential for the distribution of mitochondria into daughter cells.
Prieto et al.Citation3 recently reported that the mitochondrial fission protein Drp1 is phosphorylated early in reprogramming by ERK and that Drp1 knockdown and inhibition impair both mitochondrial fragmentation and the generation of iPSC colonies (). These data indicate that mitochondrial fission is an early and necessary step in the reprogramming process that leads to pluripotency. The activation of mitochondrial fission by phosphorylated Drp1 may precede the metabolic switch required for iPSC generation. In this issue of Cell Cycle, Prieto et al.Citation4 provide additional data on the functional contribution of mitochondrial fission proteins to cellular reprogramming. The authors document that loss-of-function of proteins involved in this mitochondrial process such as GDAP1 or MiD51, reduced the OSKM-driven mitochondrial fragmentation characteristic of the early steps of reprogramming. In contrast, silencing of mitochondrial fission proteins such as Fis1, Mff or MiD49 did not have any impact on mitochondrial morphology, thereby suggesting that these proteins do not play a significant role in mitochondrial fission during the early steps of cellular reprogramming. These data suggest the existence of specific mechanisms of mitochondrial fission during cell reprogramming. The authors further analyzed the impact of GDAP1 silencing and found a large reduction in OSKM-induced cell reprogramming (). Interestingly, the expression of GDAP1 was very low during the late steps of reprogramming, which suggests that under these conditions mitochondrial fission is independent of the fusion protein. GDAP1 loss-of-function was also associated with changes in gene expression, and the major gene pathways dysregulated were related to cell division, protein catabolic processes, and RNA processing.
The analysis of mitochondrial fusion proteins is fully coherent with current data linking mitochondrial fission and cell reprogramming. Thus, Mitofusin 1 and 2 loss-of-function facilitates the early steps of cell reprogramming induced by OSKM factors (). Cell reprogramming induced by Mfns depletion occurs through activation of HIF-1α and the increase of glycolysis, thereby indicating that modification of the mitochondrial architecture leads to the activation of the gene networks required to sustain cell reprogramming.Citation5 The molecular mechanisms that link mitochondrial dynamics and cell reprogramming remain largely unknown. It could be hypothesized that an increase in mitochondrial fission sustains undifferentiated phenotype, whereas mitochondrial fusion promotes cell differentiation. In support of this notion, Mfn2 is required for the neurogenesis and synapse formation of hiPSC-derived cortical neurons.
An additional aspect that may be of interest is the observation that GDAP1, Drp1 and Mff also participate in the fission of peroxisomes, organelles with a key catabolic function in very long chain fatty acids, amino acids, polyamines, and ROS removal. Given these considerations, peroxisomes may also play a key role in cell reprogramming. In this regard, a dominant negative form of Drp1 promotes the elongation of both mitochondria and peroxisomes, as well as lactic acidosis and increased serum levels of very long-chain fatty acids.Citation6 Further research is required to explore whether peroxisomal architecture is also relevant in the triggering of a metabolic switch during cell reprogramming.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 2011; 14:264-71; PMID:21803296; http://dx.doi.org/10.1016/j.cmet.2011.06.011
- Folmes CD, Terzic A. Energy metabolism in the acquisition and maintenance of stemness. Seminars Cell Dev Biol 2016; 52:68-75; PMID:26868758; http://dx.doi.org/10.1016/j.semcdb.2016.02.010
- Prieto J, León M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, López-García C, Torres J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun 2016; 7:11124; PMID:27030341; http://dx.doi.org/10.1038/ncomms11124
- Prieto J, León M, Ponsoda X, García-García F, Bort R, Serna E, Barneo-Muñoz M, Palau F, Dopazo J, López-García C, et al. Dysfunctional mitochondrial fission impairs cell reprogramming. Cell Cycle 2016; 15(23):3240-3250; PMID: 27753531; http://dx.doi.org/10.1080/15384101.2016.1241930
- Son,MJ, Kwon Y, Son MY, Seol B, Choi HS, Ryu SW, Choi C, Cho YS. Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ 2015; 22:1957-69; PMID:25882047; http://dx.doi.org/10.1038/cdd.2015.43
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. The N Eng J Med 2007; 356:1736-41; PMID:17460227; http://dx.doi.org/10.1056/NEJMoa064436