
Pediatric brain tumors are the leading cause of disease related death in children with pediatric low-grade gliomas (PLGGs) as the most commonly diagnosed sub-type. While surgery remains the mainstay of PLGG treatment, several patients can have non-resectable and/or disseminated tumors lead to significant tumor related morbidity. Furthermore, these slow-growing tumors are often challenging to treat with traditional chemotherapy and result in long-term neuro-toxicities in the context of the developing brain. Recently, others and we have sought to identify novel precision medicine approaches through more comprehensive profiling of the underlying mechanisms for PLGGs. In our most recent Nature Genetics publication,Citation1 we have focused on the rare PLGG subtype of angiocentric gliomas.
PLGGs are largely dominated by gene fusions, specifically BRAF fusions that aberrantly activate the mitogen-associated protein kinase (MAPK) pathway.Citation2 These findings combined with the emergence of novel MAPK therapeutics have led to clinical trials targeting the MAPK pathway in PLGGs. However, additional gene fusions found in PLGGs have remained poorly characterized. In a comprehensive evaluation of 249 PLGG patient samples, including the largest cohort of 19 AGs analyzed to date, we found a unique association between a single gene driver rearrangement, MYB-QKI and angiocentric gliomas (AGs) and sought to define its oncogenic mechanisms.
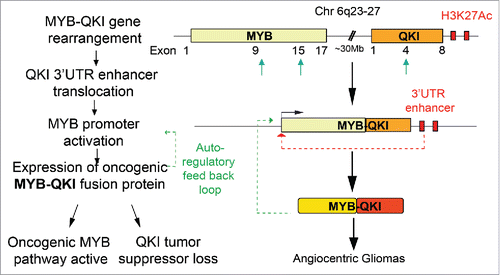
MYB-QKI occurs due to an intra-chromosomal rearrangement/deletion event on chromosome 6 where the 5′ end of MYB is fused to the 3′ end of an RNA binding protein-coding gene, QKI. MYB has been studied extensively as a proto-oncogene in hematological malignancies and solid tumors and is often activated as an oncogene by mutations and/or truncations. QKI (Quaking) is an RNA binding protein known to have essential roles in CNS development such as regulation of myelination. Recently, QKI has been suggested to be a tumor suppressor gene in several adult cancers, potentially through its regulation of microRNA functionality. We hypothesized that MYB-QKI could be driving tumorigenesis in AGs via collaborative effects of gain-of-function truncations in MYB and loss-of-function truncation of QKI. Additionally, since MYB is not normally expressed in the developed brain regions, this hinted at potential epigenetic mechanisms led by the gene rearrangements that could be altering the enhancer landscape associated with QKI.
After confirmation of MYB-QKI's capacity to promote oncogenic transformation in heterologous models, we verified that MYB-QKI has enhanced MYB-mediated transcriptional activation and, in ChIPseq assays confirmed MYB-QKI bound target genes in AGs. MYB-QKI was also found to bind to its own MYB promoter, suggestive of a positive feedback loop driving enhanced MYB-QKI expression. Analysis of the H3K27 acetylation marks around MYB-QKI revealed that in addition to this feedback mechanism, the gene rearrangement brings enhancers downstream of QKI to be newly proximal to the MYB promoter, activating the otherwise quiescent MYB promoter.
Using functional reporter assays, we demonstrated that these enhancers could activate the MYB-promoter to drive downstream MYB-QKI expression and that there is synergy between MYB-QKI driving its own promoter. These findings define a novel context for the role of epigenetic dysregulation as a result of gene fusion or other structural rearrangement events in cancers. In a companion paper by Bradley Bernstein's group reported in Nature Genetics,Citation3 similar super-enhancer translocations driven by chromosomal rearrangement events were found to activate MYB in adenoid cystic carcinomas (ACCs).
Unlike MYB, QKI's role in cancers is still emerging. In neurosphere model systems, we confirmed QKI's role as a tumor suppressor and found that QKI haploinsufficiency enhances MYB-QKI driven oncogenic phenotype while QKI loss of function alone was not sufficient to drive tumorigenesis. Remarkably, in addition to MYB-QKI fusions, others and we identified additional QKI fusion events in PLGGs, including QKI-RAF1 and QKI-NTRK2. However, in QKI-RAF1/NTRK2, the N terminus of QKI is fused to the activated RAF1/NTRK2 oncogene. The novel association of QKI with pediatric low-grade tumors hints at yet to be described co-operative mechanism by which this tumor suppressor could be functioning. QKI has been found to regulate alternative splicing,Citation4 circular RNA formation,Citation5 and microRNA processing.Citation6 Further characterization of its role in other fusion settings is needed to elucidate how such processes might impinge on PLGG biology.
Our study thus identifies a novel tripartite oncogenic mechanism being driven by a single gene arrangement in PLGGs. The gene fusion event invokes (1) MYB oncogenic activation, (2) translocation of epigenetic elements, and (3) loss of QKI's tumor suppressor functions () and highlights how simple structural rearrangements engage complex biologic mechanisms that is likely to be found in other fusion settings across cancer. This study has several important clinical implications. In the context of ongoing clinical trials employing MAPK targeting, our characterization of MYB-QKI fusions highlights the need for molecular subtyping of tumors in order to employ precision approaches based on mutational context of the tumor. While clinically targeting transcription factors remains difficult, our finding that enhancer translocation drives MYB-QKI expression suggests epigenetic-directed therapies may provide novel therapeutic opportunitiesCitation7 in the context of these fusions. Lastly, our comprehensive investigation of a rare pediatric cancer also highlights how findings in such rare diseases can also provide for a more robust understanding of likely shared mechanisms in more common adult cancers.
Figure 1. MYB-QKI gene fusions drive tri-partite oncogenic mechanisms in Angiocentric Gliomas. Schematic showing the MYB-QKI gene rearrangement event that results in enhancer translocation and expression of MYB-QKI fusion oncoprotein. MYB-QKI also drives it own expression while mediating MYB gain-of-function and QKI partial loss-of-function.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Bandopahyay P, Ramkissoon LA, Jain P, Bergthold G, Wala, J, Zeid R, Schumacher SE, Urbanski L, O'Rourke R, Gibson, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016; 48(3):273-82; PMID:26829751; http://dx.doi.org/10.1038/ng.3500
- Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, et al. R. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 2008; 118(5):1739-49; PMID:18398503; http://dx.doi.org/10.1172/JCI33656
- Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Afrogheh AH, Faquin WC. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet 2016; 48(3):265-72; PMID:26829750; http://dx.doi.org/10.1038/ng.3502
- Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni, R, and Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 2015; 43(10):5130-44; PMID:25908786; http://dx.doi.org/10.1093/nar/gkv210
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, and Goodall GJ. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015; 160:1125-34; PMID:25768908; http://dx.doi.org/10.1016/j.cell.2015.02.014
- Ji S, Ye G, Zhang J, Wang L, Wang T, Wang, Z, Zhang T, Wang G, Guo Z, Luo Y, et al. miR-574-5p negatively regulates Qki6/7 to impact β-catenin/Wnt signalling and the development of colorectal cancer. Gut 2013; 62:716-26; PMID:22490519; http://dx.doi.org/10.1136/gutjnl-2011-301083
- Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher, SE, Zeid R, Masoud S, et al. BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma. Clin Cancer Res 2014; 20:912-25; PMID:24297863; http://dx.doi.org/10.1158/1078-0432.CCR-13-2281