
ABSTRACT
The canonical process of activation of heterotrimeric G proteins by G protein coupled receptors (GPCRs) is well studied. Recently, a rapidly emerging paradigm has revealed the existence of a new, non-canonical set of cytosolic G protein modulators, guanine exchange modulators (GEMs). Among G proteins regulators, GEMs are uniquely capable of initiating pleiotropic signals: these bifunctional modulators can activate cAMP inhibitory (Gi) proteins and inhibit cAMP-stimulatory (Gs) proteins through a single short evolutionarily conserved module. A prototypical member of the GEM family, GIV/Girdin, integrates signals downstream of a myriad of cell surface receptors, e.g., growth factor RTKs, integrins, cytokine, GPCRs, etc., and translates these signals into G protein activation or inhibition. By their pleiotropic action, GIV and other GEMs modulate several key pathways within downstream signaling network. Unlike canonical G protein signaling that is finite and is triggered directly and exclusively by GPCRs, the temporal and spatial features of non-canonical activation of G protein via GIV-family of cytosolic GEMs are unusually relaxed. GIV uses this relaxed circuitry to integrate, reinforce and compartmentalize signals downstream of both growth factors and G proteins in a way that enables it to orchestrate cellular phenotypes in a sustained manner. Mounting evidence suggests the importance of GIV and other GEMs as disease modulators and their potential to serve as therapeutic targets; however, a lot remains unknown within the layers of the proverbial onion that must be systematically peeled. This perspective summarizes the key concepts of the GEM-dependent G protein signaling paradigm and discusses the multidisciplinary approaches that are likely to revolutionize our understanding of this paradigm from the atomic level to systems biology.
Canonical heterotrimeric G protein signaling; signals are initiated by GPCRs and ‘adjusted’ by accessory proteins
Heterotrimeric G proteins are major signaling hubs and essential components of the signal gating machinery in healthy eukaryotic cells. In the canonical scenario, activation of G proteins is triggered by agonist-bound GPCRs. By directly coupling to the Gα-subunit of the αβγ heterotrimer,Citation1-4 GPCRs induce a conformational change leading to loss of bound GDP and dissociation of Gβγ subunits; in other words, GPCRs serve as guanine nucleotide exchange factors (GEFs; []). The unparalleled importance of G protein signaling has been recognized for a long time and is reflected in the fact that ∼40% of currently marketed drugs target GPCRs.Citation5
Figure 1. Mechanism of action of GEMs. Schematic comparing and contrasting key features of canonical (GPCR-triggered, left) and non-canonical (GEM-triggered, right) heterotrimeric G protein signaling. Left: Canonical activation of heterotrimeric G proteins is spatially and temporally restricted, i.e., triggered exclusively by agonist activated G-protein-coupled receptors (GPCRs) (signal input), may either lead to inhibition or stimulation of adenylate cyclase and cAMP production (signal output) depending on whether inhibitory Giαβγ or stimulatory Gsαβγ heterotrimers are activated, respectively. Such activation is triggered primarily at the plasma membrane (PM) via a finite process that is rapid and completes within a few hundred milliseconds, and may continue on PM-contiguous endocytic compartments.Citation32-34 Right: Non-canonical G protein signaling that is triggered by GEMs are characterized by 4 key differences (from top to bottom) – (A) GEMs can coordinately modulate heterotrimeric G protein signaling downstream of diverse classes of receptors (signal input). (B) GEMs can activate monomeric Giα (as a GEF) or inhibit stimulatory Gsα (as a GDI) using the same short motif, thereby coordinately reducing cellular cAMP. The monomeric Gα substrate could either be a byproduct of GPCR signaling or actively released from Gi/sαβγ trimers by displacement of Gβγ heterodimers or GoLoco/GPR motif containing GDIs. (C) Being cytosolic in localization, GEMs can act upon heterotrimeric G proteins on both PM and internal membranes that are non-contiguous with the PM, e.g., Golgi and autophagosomes. (D) Signaling that is triggered by GEMs appears to be delayed (initiated after several minutes) and prolonged, lasting several minutes to hours (reviewed inCitation14). Note: The spatiotemporal features listed here exclusively refer to the nature of heterotrimeric G protein activation within each paradigm, and do not refer to other downstream signals initiated within each pathway (like MAPK or Akt), or downstream gene transcription/translation responses that are typically delayed, or effects on the cytoskeleton or organelles that are brought about directly or indirectly via downstream intermediates within the signaling cascades.
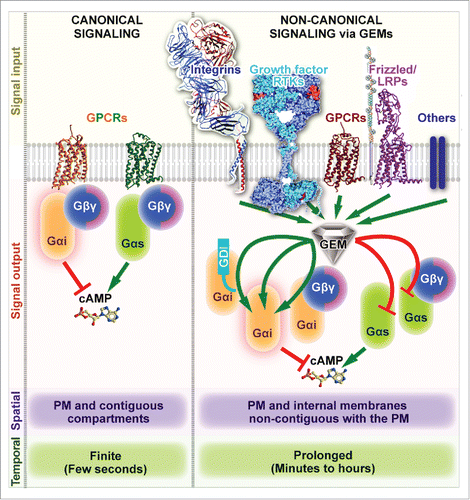
In the past few decades, it has become clear that GPCRs represent only one of the multiple access points to the signaling hub of G proteins. G protein signaling that is initiated by GPCRs is further fine-tuned by the “GAPs, GEFs and GDIs of heterotrimeric G-protein α-subunits”Citation6– a heterogeneous set of accessory proteins. Among them, GTPase accelerating proteins (GAPs) promote GTP hydrolysis to GDP by the Gα subunit, and thereby, they terminate Gα signaling, and guanine nucleotide dissociation inhibitors (GDIs) stabilize the inactive, GDP-bound state of the Gα subunits, and thereby, they inhibit activation of Gα-proteins. The members of the third group, non-receptor GEFs, are a heterogeneous group of cytosolic proteins that accelerate GDP to GTP exchange, and thereby, cause activation of Gα subunits through a non-canonical mechanism, independently of GPCRs or Gβγ dimers.Citation7-11 These fundamentals have been described in-depth in a widely-cited review by Siderovski and WillardCitation6 over a decade ago. In the years since, a growing body of work by us and others indicates that genetic or epigenetic factors that deregulate the intricate network of “accessory proteins” in a variety of disease states are just as significant as those that directly affect the G proteins and GPCRs, if not more so.Citation7,12-14
Non-canonical G protein signaling by GEMs; The serendipitous discovery of a family of proteins based on homology to a synthetic peptide
The wide prevalence and broad significance of deregulated G protein regulatory network in diseases (e.g., multiple cancers, type II diabetes and organ fibrosisCitation14) is perhaps best exemplified by the recently identified family of Guanine-nucleotide Exchange Modulators (GEMs).Citation15 The members of this family share very little sequence homology and act within diverse signaling cascades; what unites them is the ability to couple activation of these cascades to G protein signaling () via an evolutionarily conserved motif of ∼30 aa that directly binds to G proteins.
The prototypical member of the GEM family, a multi-domain cytosolic protein GIV (a.k.a Girdin, HkRP1 and APE) was co-discovered independently by 4 groups in 2005–06. As it often happens in biology, the discovery process was reminiscent of the ancient fableCitation16 in which several blind men provide seemingly conflicting descriptions of the same elephant, depending on the body part of the elephant each of them could reach. For example, in 2005, Le-Niculescu et al.Citation17 identified this protein as a binding partner for Gαi and Gαs via yeast 2-hybrid display, and found that it localizes on vesicles at/near the Golgi, justifying the name of GIV (Gα-interacting Vesicle-associated). Three other groups co-discovered GIV nearly simultaneously. Anai et alCitation18 described it as a binding partner and activator of Akt kinase and named it APE (Akt-Phosphorylation Enhancer). Simpson et al.Citation19 reported it as a Hook-domain containing microtubule-interacting protein which binds the large GTPase dynamin and modulates endocytic trafficking; they named it HkRP1 (Hook-Related Protein 1). Finally, Enomoto et al.Citation20 described it as a cytoskeleton-binding protein that remodels actin during cell migration and named it Girdin, for “GIRDer of actIN.” Of the multiple interesting functional modules in GIV, the one allowing it to bind to G proteins turned out to be pivotal for GIV's role in cell migrationCitation21; however, the molecular basis of this interaction remained unclear.
That same year (i.e., 2005), in a seemingly unrelated work, Johnston et al.Citation22 searched for guanine nucleotide-selective Gα binding peptides using the technique of phage display, and found a short (16 aa) synthetic peptide, KB-752, that not only exclusively associated with the GDP-bound Gαi but also enhanced spontaneous nucleotide exchange, i.e., possessed GEF functionality on Gαi. Later that year, the same group demonstrated that KB-752 also binds Gαs•GDPCitation23; however, for this G protein, the peptide acted as a GDI, i.e., it slowed the rate of nucleotide exchange. In other words, KB-752 demonstrated unique bifunctional behavior with opposing action on Gαi and Gαs. Unfortunately, at the time of publication, proteome-wide sequence searches failed to detect significant homology of KB-752 to any naturally-occurring sequence.
Within the decade that followed, GIV and a few other Gα-binding proteins (identified through the same yeast-2-hybrid assay as GIVCitation17) were shown to contain a sequence motif with distant homology to KB-752. For GIV, such homology was reported in 2009. Calnuc/Nucleobindin 1 and 2 (NUCB1 and NUCB2) followed in 2011,Citation24 and Daple in 2015.Citation25 All these proteins were shown to specifically bind the GDP-bound Gαi-subunit and enhance nucleotide exchange using the same KB-752-like evolutionarily conserved motif. Consequently, they all serve as non-receptor GEFs for Gαi. In the case of GIV, Gupta V., et alCitation15 recently demonstrated that as KB-752, GIV is a GDI for GαsCitation15: it inhibits the Gαs→cAMP→PKA→pCREB signaling pathway in cells using the same KB-752-like motif. These findings exposed the need for a new nomenclature, i.e., “GEM,” primarily to distinguish the KB-752-like family of proteins from the other non-receptor GEFs (like Ric8A/B or AGS1) that have no propensity for pleiotropy or dual (Gαi activating/Gαs inhibitory) function.
As summarized above, GIV is the first and the most rigorously investigated member of the GEM family.Citation26 Several studiesCitation26-28 have provided structural insights into the assembly of the GIV-Gαi complex via a combination of homology modeling (using the structure of Gαi-bound KB-752 peptide as templateCitation22) and site-directed mutagenesis. These studies revealed that the KB-752-like GEM motif of GIV engages a hydrophobic cleft between the switch II and the α3 helix of Gαi, a mechanism distinct from how Gα-subunits engage with GPCRs.Citation2 Such a mode of binding explains why GIV-GEM cannot bind active GTP-associated Gαi (the conformation of Gαi switch II is nucleotide-dependent and sterically incompatible with GIV binding in the GTP-associated stateCitation29), and provides clues as to how GIV activates monomeric Gαi, either directly or after competitively displacing from Gαi both GoLoco/GPR motif containing GDIsCitation27 as well as the canonical GDI GβγCitation26 (). These key structural insights led to identification of a selective GEF/GDI-deficient GIV mutant (F1685A) that proved to be a powerful and precise tool for dissecting the biologic roles and unraveling the spatiotemporal features of G protein signaling that is triggered by GIV. Using this mutant, multiple studies (summarized inCitation30) have demonstrated that the signaling network triggered in cells with wild-type GIV is a mirror image of the network in cells lacking GIV's GEF/GDI functionality: signals that are enhanced in cells that are GEF/GDI-proficient are suppressed in cells that are GEF/GDI-deficient, and vice versa. Our understanding of the spatiotemporal aspects and the impact of all known GEMs is still incomplete; however, studies on GIV made it clear that GEM-triggered G protein signaling is governed by a new set of rules as compared with canonical GPCR-dependent signalingCitation30 (). First, the rules of receptor engagement are different. Unlike the canonical pathway, where heterotrimeric G proteins engage exclusively with ligand-activated receptor GPCRs, the non-receptor GIV-GEF engages with a diverse array of receptors, including GPCRs, Integrins and RTKs.Citation26,30,31 and, thereby, enables transactivation of G proteins in response to a wide variety of stimuli (). Second, the temporal aspects of G protein signaling are distinct; canonical GPCR-dependent G protein signaling is short-lived (lasts seconds), whereas signaling via GIV-GEM is delayed and prolonged (lasts several min) (). Third, the spatial pattern of heterotrimeric G protein activation by GIV also poses a stark contrast to canonical heterotrimeric G protein signaling. Canonical G protein activation by ligand-activated GPCRs is initiated exclusively at the PM and may continue on PM-contiguous endocytic compartmentsCitation32-34 By contrast, non-canonical heterotrimeric G protein activation by GIV-GEM can be triggered at multiple intracellular compartments, including centrosomes, focal adhesions, cell-cell junctions, early endosomes, exocytic vesicles, autophagosomes, and more recently, on Golgi membranes (summarized inCitation30) (). These unique characteristics make the entire paradigm of non-canonical heterotrimeric G protein signaling via GEMs fundamentally different and novel.
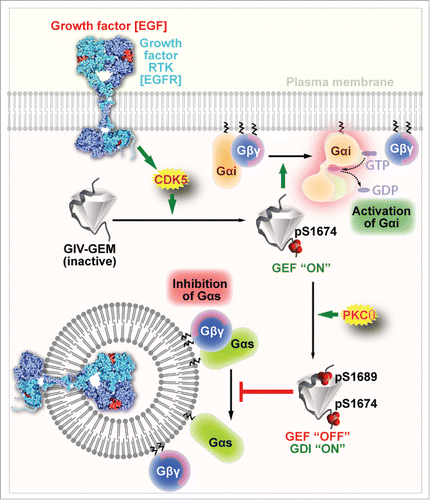
Gupta V., et al.,Citation15 also revealed how the pleiotropic GEF/GDI functions of GEMs may be controlled by post-translational modifications. They showed that in the case of the prototypical GEM GIV, sequential phosphorylation of 2 Ser residues that flank the bifunctional GEF/GDI motif on GIV by 2 kinases, CDK5Citation35 and PKCθ,Citation36 ensures that GIV exerts its GEF and GDI activities on Gαi and Gαs, respectively, in a temporally and spatially segregated manner (). Such temporal-spatial regulation appears to integrate, reinforce and compartmentalize signals downstream of both growth factors and G proteins and dictate whether cells migrate or divide. Whether or not the other known members of the GEM family also function as GDIs for Gαs, and if so, what post-translational modifications may regulate such function remains to be determined.
Figure 2. Key phosphoevents regulate the spatiotemporally separated GEF and GDI functions of GIV. Schematic showing the spatially separated GEF and GDI actions of GIV-GEM on Gαi and Gαs, respectively. Upon EGF stimulation, activated CDK5 kinase phosphorylates GIV on S1674Citation35, thereby turning ‘on’ its GEF function toward Gαi. Subsequently, GIV is phosphorylated also at S1689 by PKCθCitation36; this phosphoevent turns ‘off’ GIV's GEF function, but turns ‘on’ its GDI function toward Gαs. Such coordinated activation of Gαi first at the PM within 5 min after EGF stimulation and subsequent inhibition of Gαs on endosomes at ∼15–30 min after EGF stimulation ensures that cAMP levels are suppressed both early and late in cells responding to EGF.
Last, but not least, using an unbiased bioinformatics search in Caenorhabditis elegans Coleman et al.Citation37 have recently identified GBAS-1 (GBA and SPK domain containing-1) as a KB-752-like motif-containing protein with homologs only in closely related worm species and demonstrates that GBAS-1 has GEF activity for the cognate Gα in C. elegans, i.e., GOA-1. Their findings underscore the possibility that GEMs, and the paradigm of non-canonical G protein signaling they trigger, may mediate receptor-independent G protein activation also in metazoans.
Pleiotropic heterotrimeric G protein signaling triggered by GEMs coordinate suppression of a key universal second messenger, cyclic AMP
Ca2+ and cAMP are recognized as universal regulators of cell function. Between them, these 2 second messengers impact nearly every aspect of cellular life in diverse organisms ranging from single-cell organisms to humans. Confinement of Ca2+ or cAMP to precise subcellular domains (e.g., plasma membrane, organelles) allows these molecules to selectively activate a subset of targets, thereby expanding the repertoire and range of the signal. Although the effect of GEMs on Ca2+ is still under investigation, the effect of GEMs on cAMP is clear. GEMs suppress cellular cAMP by exerting opposing effects on 2 classes of heterotrimeric G proteins that have antagonistic effects on adenylyl cyclase (AC); they activate the inhibitor of AC, i.e., Gi and inhibit the stimulator of AC, i.e., Gs. Because activation of Gi and inhibition of Gs by GIV occurs in spatially and temporally segregated manner, such segregation may enable compartmentalization of cAMP pulses. These insights provide new clues into how GIV and other GEMs may achieve sustained and coordinated cAMP signals within various cellular compartments. Because tight regulation of the second messenger cAMP is of crucial importance in both health and disease,Citation38 it is not surprising that GIV's GEM function has been found to be essential for a variety of cellular processes in physiology and its dysregulation (expression or function) a contributing factor in a variety of disease states (reviewed inCitation14).
‘Free’ Gβγ-Heterodimers are major intermediates in pleiotropic G protein signaling triggered by GEMs
By triggering activation of Gi and inhibition of Gs, GIV-GEM accomplishes another final common goal, i.e., release of ‘free’ Gβγ and activation of PI3K-Akt signals, thereby integrating and reinforcing the strength of the final signaling response.Citation15,26 Using Gallein,Citation26 a compound that blocks Gβγ-interactions with PI3Kγ by binding to a protein-protein interaction “hot spot” on the Gβ subunit,Citation39 it was demonstrated that the mechanism of PI3K-Akt enhancement brought about by GIV's GEM motif involves the release of ‘free’ Gβγ-heterodimers from both Gi and Gs trimers. Because Gβγ subunits interact with other components of canonical G protein signaling complexes (summarized inCitation40), e.g., receptors, Gα subunits, effectors (including GAPs and GEFs for small GTPases), and regulatory enzymes such as G protein–coupled receptor kinases (GRKs), it is clear that GEMs may influence several aspects of signaling that are fine-tuned by Gβγ subunits. Finally, it should be noted that because all Gβγ dimer combinations are not equal, those displaced from Gi at the PM may have distinct functions from those displaced from Gs on endosomes. Different cellular pools of Gβγ released at different locations are likely to control a great deal of the architecture of cellular signaling that is triggered by GEMs.
Future directions
Among a long list of unanswered questions in this rapidly evolving paradigm, 2 fields/disciplines hold the greatest promise to deliver the most insights in the immediate future. First, crystallographic elucidation of the structural basis for the pleiotropic GEF/GDI action of a single short functional motif in GEMs on Gαi and Gαs, respectively, will go a long way in legitimizing this new family of modulators. As crystal structures and biophysical studies have revolutionized our understanding of canonical, GPCR-triggered G protein signaling (), crystal structures of Gα:GEM complexes may do the same for non-canonical signaling. Together with the existing structures documenting the GDI and GAP action of well-defined signature motifs/domains like GoLoco or RGS-box, the future Gα:GEM structures will resolve the picture of heterotrimeric G protein regulation at an atomic level and will undoubtedly guide the development of therapeutics targeting specific interactions and steps within this completed picture. Efforts are underway to gain atomic level insights into the mechanism of action of GEMs, a need that is both unmet and urgent. Structural insights are also expected to define the essential/invariable requirements of a GEF/GDI bifunctional motif and thereby, enable the discovery of other GEMs in the human genome.
Second, although growth factor-dependent G protein signaling may appear to be a linear connection between input (the growth factor receptors) and output (G-proteins) elements, experimental data shows that GEMs like GIV are rather an integral part of a network that links many receptors to many signaling pathways, and links multiple cellular organelles to events at the PM.Citation14 The behavior of such complex systems is hard to grasp by intuition. Systems biology approaches, particularly mathematical and computational modeling, have emerged as an important toolkit for studying these signaling pathways.Citation41,42 Because multiple feedforward/feedback cycles modulate GIV-dependent signaling and orchestrate it in separate time and space (), mathematical modeling, constrained by experimental data, is expected to be more reliable. Such an approach will help generate more comprehensive models to illuminate how GIV drives diverse cellular processesCitation14 in a spatio-temporal manner and lead to a mechanistic and predictive framework for experimental design. Ongoing efforts are underway to build such a framework that will integrate experimental knowledge into a coherent picture so we can test, support, or falsify our hypotheses of mechanism of action of GEMs.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by NIH (R01CA160911, R01CA100768 and DK099226) to P.G.
References
- Dror RO, Mildorf TJ, Hilger D, Manglik A, Borhani DW, Arlow DH, Philippsen A, Villanueva N, Yang Z, Lerch MT, et al. SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 2015; 348:1361-1365; PMID:26089515; http://dx.doi.org/10.1126/science.aaa5264
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011; 477:549-555; PMID:21772288; http://dx.doi.org/10.1038/nature10361
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 2011; 469:236-240; PMID:21228876; http://dx.doi.org/10.1038/nature09665
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011; 469:175-180; PMID:21228869; http://dx.doi.org/10.1038/nature09648
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002; 1:727-730; PMID:12209152; http://dx.doi.org/10.1038/nrd892
- Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci 2005; 1:51-66; PMID:15951850; http://dx.doi.org/10.7150/ijbs.1.51
- Sato M, Blumer JB, Simon V, Lanier SM. Accessory proteins for G proteins: partners in signaling. Annu Rev Pharmacol Toxicol 2006; 46:151-187; PMID:16402902; http://dx.doi.org/10.1146/annurev.pharmtox.46.120604.141115
- Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci 2005; 1:51-66; http://dx.doi.org/10.7150/ijbs.1.51
- Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 2000; 69:795-827; PMID:10966476; http://dx.doi.org/10.1146/annurev.biochem.69.1.795
- Blumer JB, Oner SS, Lanier SM. Group II activators of G-protein signalling and proteins containing a G-protein regulatory motif. Acta Physiol (Oxf) 2012; 204:202-218; PMID:21615707; http://dx.doi.org/10.1111/j.1748-1716.2011.02327.x
- De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG. The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol 2000; 40:235-271; PMID:10836135; http://dx.doi.org/10.1146/annurev.pharmtox.40.1.235
- Sjogren B. Regulator of G protein signaling proteins as drug targets: current state and future possibilities. Adv Pharmacol 2011; 62:315-347; PMID:21907914
- Appleton KM, Bigham KJ, Lindsey CC, Hazard S, Lirjoni J, Parnham S, Hennig M, Peterson YK. Development of inhibitors of heterotrimeric Galphai subunits. Bioorg Med Chem 2014; 22:3423-3434; PMID:24818958; http://dx.doi.org/10.1016/j.bmc.2014.04.035
- Aznar N, Kalogriopoulos N, Midde KK, Ghosh P. Heterotrimeric G protein signaling via GIV/Girdin: Breaking the rules of engagement, space, and time. Bioessays 2016; 38:379-393; PMID:26879989; http://dx.doi.org/10.1002/bies.201500133
- Gupta V, Bhandari D, Leyme A, Aznar N, Midde KK, Lo IC, Ear J, Niesman I, Lopez-Sanchez I, Blanco-Canosa JB, et al. GIV/Girdin activates Galphai and inhibits Galphas via the same motif. Proc Natl Acad Sci U S A 2016; 113:E5721-5730; PMID:27621449; http://dx.doi.org/10.1073/pnas.1609502113
- Yankelowitz BY. Biology, blind men, and elephants. Br Med J 1978; 2:1775; PMID:20792776; http://dx.doi.org/10.1136/bmj.2.6154.1775
- Le-Niculescu H, Niesman I, Fischer T, DeVries L, Farquhar MG. Identification and characterization of GIV, a novel Galpha i/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J Biol Chem 2005; 280:22012-22020; PMID:15749703; http://dx.doi.org/10.1074/jbc.M501833200
- Anai M, Shojima N, Katagiri H, Ogihara T, Sakoda H, Onishi Y, Ono H, Fujishiro M, Fukushima Y, Horike N, et al. A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J Biol Chem 2005; 280:18525-18535; PMID:15753085; http://dx.doi.org/10.1074/jbc.M500586200
- Simpson F, Martin S, Evans TM, Kerr M, James DE, Parton RG, Teasdale RD. Wicking CA novel hook-related protein family and the characterization of hook-related protein 1. Traffic 2005; 6:442-458; PMID:15882442; http://dx.doi.org/10.1111/j.1600-0854.2005.00289.x
- Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell 2005; 9:389-402; PMID:16139227; http://dx.doi.org/10.1016/j.devcel.2005.08.001
- Ghosh P, Garcia-Marcos M, Bornheimer SJ, Farquhar MG. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol 2008; 182:381-393; PMID:18663145; http://dx.doi.org/10.1083/jcb.200712066
- Johnston CA, Willard FS, Jezyk MR, Fredericks Z, Bodor ET, Jones MB, Blaesius R, Watts VJ, Harden TK, Sondek J, et al. Structure of Galpha(i1) bound to a GDP-selective peptide provides insight into guanine nucleotide exchange. Structure 2005; 13:1069-1080; PMID:16004878; http://dx.doi.org/10.1016/j.str.2005.04.007
- Johnston CA, Ramer JK, Blaesius R, Fredericks Z, Watts VJ, Siderovski DP. A bifunctional Galphai/Galphas modulatory peptide that attenuates adenylyl cyclase activity. FEBS Lett 2005; 579:5746-5750; PMID:16225870; http://dx.doi.org/10.1016/j.febslet.2005.09.059
- Garcia-Marcos M, Kietrsunthorn PS, Wang H, Ghosh PM, Farquhar MG. G Protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of G(alpha)i-regulatory motifs. J Biol Chem 2011; 286:28138-28149; PMID:21653697; http://dx.doi.org/10.1074/jbc.M110.204099
- Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, Barbazan J, Murray F, Nitsche U, Janssen KP, et al. Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. Elife 2015; 4:e07091; PMID:26126266; http://dx.doi.org/10.7554/eLife.07091
- Garcia-Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc Natl Acad Sci U S A 2009; 106:3178-3183; PMID:19211784; http://dx.doi.org/10.1073/pnas.0900294106
- Garcia-Marcos M, Ear J, Farquhar MG, Ghosh P. A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol Biol Cell 2011; 22:673-686; PMID:21209316; http://dx.doi.org/10.1091/mbc.E10-08-0738
- Garcia-Marcos M, Ghosh P, Ear J, Farquhar MG. A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. J Biol Chem 2010; 285:12765-12777; PMID:20157114; http://dx.doi.org/10.1074/jbc.M109.045161
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG¸Sprang SR. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science (New York, N.Y) 1994; 265:1405-1412; PMID:8073283; http://dx.doi.org/10.1126/science.8073283
- Aznar N, Kalogriopoulos N, Midde K, Lo IC¸Ghosh P. Heterotrimeric G protein signaling via GIV/Girdin: Breaking the rules of engagement, space and time. BioEssays 2016; 38(4):379-93; PMID:26879989
- Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, Ear J, Jung BH, Cabrera B, Carethers JM, et al. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell 2010; 21:2338-2354; PMID:20462955; http://dx.doi.org/10.1091/mbc.E10-01-0028
- Tsvetanova NG, Irannejad R, von Zastrow M. G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem 2015; 290:6689-6696; PMID:25605726; http://dx.doi.org/10.1074/jbc.R114.617951
- Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol 2014; 27:109-116; PMID:24680436; http://dx.doi.org/10.1016/j.ceb.2013.10.003
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013; 495:534-538; PMID:23515162; http://dx.doi.org/10.1038/nature12000
- Bhandari D, Lopez-Sanchez I, To A, Lo IC, Aznar N, Leyme A, Gupta V, Niesman I, Maddox AL, Garcia-Marcos M, et al. Cyclin-dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration-proliferation dichotomy. Proc Natl Acad Sci U S A 2015; 112:E4874-4883; PMID:26286990; http://dx.doi.org/10.1073/pnas.1514157112
- Lopez-Sanchez I, Garcia-Marcos M, Mittal Y, Aznar N, Farquhar MG, Ghosh P. Protein kinase C-theta (PKCtheta) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proc Natl Acad Sci U S A 2013; 110:5510-5515; PMID:23509302; http://dx.doi.org/10.1073/pnas.1303392110
- Coleman BD, Marivin A, Parag-Sharma K, DiGiacomo V, Kim S, Pepper JS, Casler J, Nguyen LT, Koelle MR, Garcia-Marcos M. Evolutionary Conservation of a GPCR-Independent Mechanism of Trimeric G Protein Activation. Mol Biol Evol 2016; 33:820-837; PMID:26659249; http://dx.doi.org/10.1093/molbev/msv336
- Zaccolo M. Spatial control of cAMP signalling in health and disease. Curr Opin Pharmacol 2011; 11:649-655; PMID:22000603; http://dx.doi.org/10.1016/j.coph.2011.09.014
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of Gbetagamma-subunit signaling with small molecules. Science 2006; 312:443-446; PMID:16627746; http://dx.doi.org/10.1126/science.1120378
- Dupre DJ, Robitaille M, Rebois RV, Hebert TE. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol 2009; 49:31-56; PMID:18834311; http://dx.doi.org/10.1146/annurev-pharmtox-061008-103038
- Azeloglu EU, Iyengar R. Signaling networks: information flow, computation, and decision making. Cold Spring Harb Perspect Biol 2015; 7:a005934; PMID:25833842; http://dx.doi.org/10.1101/cshperspect.a005934
- Ma'ayan A, Iyengar R. From components to regulatory motifs in signalling networks. Brief Funct Genomic Proteomic 2006; 5:57-61; PMID:16769680; http://dx.doi.org/10.1093/bfgp/ell004