
ABSTRACT
Hepatocellular carcinoma (HCC), the most frequent primary tumor of the liver, is an aggressive cancer type with limited treatment options. Cumulating evidence underlines a crucial role of aberrant lipid biosynthesis (a process known as de novo lipogenesis) along carcinogenesis. Previous studies showed that suppression of fatty acid synthase (FASN), the major enzyme responsible for de novo lipogenesis, is highly detrimental for the in vitro growth of HCC cell lines. To assess whether de novo lipogenesis is required for liver carcinogenesis, we have generated various mouse models of liver cancer by stably overexpressing candidate oncogenes in the mouse liver via hydrodynamic gene delivery. We found that overexpression of FASN in the mouse liver is unable to malignantly transform hepatocytes. However, genetic deletion of FASN totally suppresses hepatocarcinogenesis driven by AKT and AKT/c-Met protooncogenes in mice. On the other hand, liver tumor development is completely unaffected by FASN depletion in mice co-expressing β-catenin and c-Met. Our data indicate that tumors might be either addicted to or independent from de novo lipogenesis for their growth depending on the oncogenes involved. Additional investigation is required to unravel the molecular mechanisms whereby some oncogenes render cancer cells resistant to inhibition of de novo lipogenesis.
Hepatocellular carcinoma: A deadly disease with a rising incidence worldwide
The global incidence of primary liver cancer has progressively increased during the recent decades. Among primary liver malignancies, hepatocellular carcinoma (HCC) is the most prevalent: it affects approximately one million individuals annually in the world, with the incidence roughly equal to the mortality rate.Citation1-3 The occurrence of HCC is highest in men, and the discrepancy in the geographical distribution of HCC reflects the differences in exposure to hepatitis viruses and distinct environmental pathogens. The incidence is highest in East Asia and Sub-Saharan Africa, representing about 85% of the total number of cases worldwide, while in most industrialized countries HCC occurrence is low, except for Southern Europe.Citation1-3 Multiple risk factors have been linked to the development of HCC, with chronic viral hepatitis (B and C) infection, alcohol abuse, non-alcoholic steatohepatitis (NASH), presence of diabetes and/or metabolic syndrome, and exposure to aflatoxins being the most frequent; nonetheless, HCC can also occur in people without any known risk factor.Citation4-6 The clear majority of HCC patients are diagnosed with an advanced disease, often precluding potentially effective therapies such as liver transplantation or curative partial liver resection. Targeted therapies against HCC are very limited. Sorafenib, a multi-kinase inhibitor, is the only drug available for the treatment of unresectable HCC. Its approval was based on the results from a clinical trial showing an increased survival of 2–3 months in Sorafenib-treated patients.Citation7 Many other drugs, including tyrosine kinase inhibitors (TKIs) and Rapalogs, have also been tested in clinical trials, but none of them proved to be more effective than Sorafenib in HCC.Citation8-11 Thus, there is an obvious unmet medical need for new drug targets for the treatment of liver cancer.
Our understanding of the HCC pathogenesis has significantly improved during the past decade, especially due to the development of genome-wide analysis methods.Citation12 Using these techniques, several critical signaling pathways that promote HCC development and/or progression have been identified, including Wnt/β-catenin,Citation13 Jak/Stat,Citation14 Ras/MAPK,Citation15 PI3K/AKT/mTOR,Citation16 c-Met,Citation17 and EGFRCitation18 cascades. These pathways can represent potential targets for HCC therapeutics.
De novo lipogenesis in cancer
Metabolic rewiring is now recognized as one of the hallmarks of cancer.Citation19 This metabolic reprogramming goes far beyond the well-known Warburg effect and it can be observed in multiple networks of central carbon metabolism. One of the metabolic networks that is deregulated along carcinogenesis is the fatty acid synthesis pathway. Specifically, increased de novo lipogenesis is frequently observed in tumor cells.Citation20-22 De novo lipogenesis is the synthesis of endogenous fatty acids from acetyl-CoA.Citation23 It is a highly-coordinated process, which involved several lipogenic enzymes (). In brief, upon glucose entry into the cell, pyruvate is generated through the glycolytic process in the cytosol. In the mitochondria, pyruvate is then converted into citrate through the tricarboxylic acid (TCA) cycle. Citrate exits mitochondria, and is converted to acetyl-CoA by ATP citrate lyase (ACLY). Next, acetyl-CoA carboxylase (ACC) converts acetyl-CoA into malonyl-CoA via irreversible carboxylation. Finally, fatty acid synthase (FASN), the key metabolic enzyme responsible for the terminal catalytic step in fatty acids biosynthesis, induces the condensation of acetyl-CoA and malonyl-CoA to produce palmitic acid, the 16-carbon saturated fatty acid. Palmitic acid is then elongated by the elongation of very long chain fatty acids protein 6 (Elovl6) elongase and forms stearic acid. Thereafter, stearoyl-CoA desaturase 1(SCD1) is responsible for desaturation of palmitic and stearic acid on Δ9 position, resulting in the formation of the palmitoleate and oleate monounsaturated fatty acids (MUSFAs). The fatty acids can be further desaturated by Fatty acid desaturase (FADS) to form polyunsaturated fatty acids (PUSFAs). Eventually, all free fatty acids (FAs) will be used as substrates for the synthesis of triglycerides (TG), cholesterol esters (CE) and phospholipids (PL).
Figure 1. De novo lipogenesis in the cell. Abbreviations: MUSFA, monounsaturated fatty acids; PUSFA, polyunsaturated fatty acids; FADS, fatty acid desaturase; CE, cholesterol esters; TG, triglyceride; PL, phospholipid; TCA, tricarboxylic acid; ACC, acetyl-CoA carboxylase; ELOVL, elongation of very long chain fatty acids protein; FASN, fatty acid synthase; SCD1, stearoyl-CoA desaturase 1; ACLY, adenosine triphosphate citrate lyase; TCA, tricarboxylic acid. Details are reported in the main text.
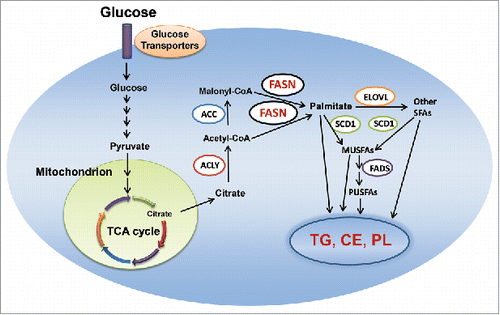
In most adult tissues, energy-providing lipids come almost exclusively from circulating (dietary) lipids, apart from the liver, adipose tissue, and lactating breast, where the rate of de novo lipogenesis is relatively high. Lipids deriving from de novo biosynthesis provide also energy supply for fetal tissues.Citation24
Importantly, mounting evidence implies a pivotal role of proteins involved in de novo lipogenesis in the development and progression of various human neoplasms, including tumors from breast, colorectal, prostate, bladder, ovary, gastrointestinal tract, lung, oral cavity, and head and neck..Citation21,22 In cancer, deregulated lipid synthesis is necessary to provide a constant source of lipids and lipid precursors to promote membrane production and lipid-based post-translational modification of proteins needed for signal transduction in a context of elevated proliferation.Citation21,22 At the molecular level, the uncontrolled de novo lipid biosynthesis is underscored by the increased activity and expression of several lipogenic enzymes in neoplastic cells, including ACLY, ACC, FASN, and SCD1. For instance, ACLY increased expression and activity have been reported in cancer cells, where small RNA interference (siRNA)-directed and pharmacological inhibition of ACLY strongly reduced the proliferation and survival of cancer cells.Citation25 Similarly, knockdown of ACAC, FASN, and SCD1 by siRNA or inhibition of ACC or FASN with small molecular inhibitors were found to impair tumor cell proliferation and induces apoptosis in various in vitro and in vivo cancer models.Citation22,26,27 Altogether, this body of information supports a pivotal role of lipogenic proteins and de novo lipid synthesis in cancer initiation and progression.
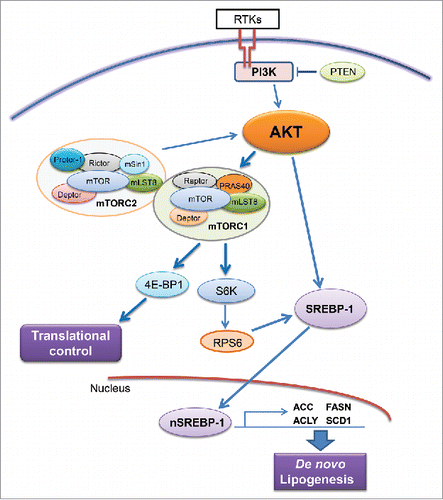
Increased de novo lipogenesis in cancer cells is regulated by multiple mechanisms.Citation21 Among them, the activated phosphoinositide 3-kinase (PI3K)/v-Akt murine thymoma viral oncogene homolog (AKT)/mammalian target of rapamycin (mTOR) cascade has been identified as a major pathway promoting lipogenesisCitation28 (). In tumor cells, PI3K is activated by the activation of receptor tyrosine kinases, such as EGFR or c-Met, loss of the tumor suppressor gene Pten or activating mutations of PIK3CA, leading to the activation of AKT. AKT in turn activates the mTOR complexes (mTORC), which include mTORC1 and mTORC2.Citation29 mTORC2 functions as feedback mechanism and is required for the activation of AKT. mTORC1 instead phosphorylates 4E-BP1, releasing its inhibitory effect on eIF4E and promoting protein synthesis. mTORC1 also phosphorylates p70S6K, leading to the phosphorylation of RPS6 and increased expression and process of sterol regulatory element binding protein1/2 (SREBP1/2). SREBPs, a family of basic-helix-loop-helix-leucine zipper transcription factors are the major regulators of lipogenesis.Citation30 Thus, increased lipogenesis is a major metabolic readout of increased AKT/mTOR signaling in tumor cells.
Figure 2. Regulation of de novo lipogenesis via the AKT/mTOR cascade. Abbreviations: RTKs, receptor tyrosine kinases; PTEN, phosphatase and tensin homolog; mTOR, mammalian target of rapamycin; mTOR, mTOR complex; SREBP-1, sterol regulatory element-binding protein-1. Details are reported in the main text.
Increased lipogenesis in HCC
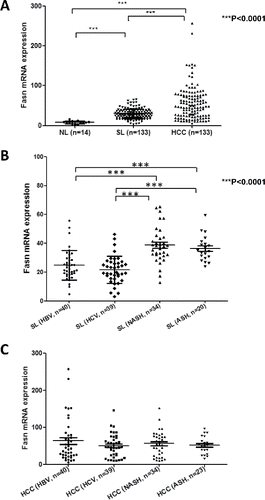
Consistent with other tumor types, increased expression of lipogenic pathway genes, including FASN, has been reported in human HCC.Citation31-33 In accordance with previous data, we have found a progressive upregulation of FASN mRNA from human normal livers to HCC in our sample collection (). Of note, our preliminary data show that levels of FASN are higher in surrounding non-tumorous livers of HCC associated with the presence of NASH and alcoholic steatohepatitis (ASH) than in surrounding livers of HCC associated with HBV and HCV chronic infection (). However, this etiology-related difference in FASN expression is not detectable in the tumor counterpart (). These findings suggest that FASN might play a more or less pronounced role depending of the etiology associated with HCC development, at least in the early phases of hepatocarcinogenesis. However, a more comprehensive study is necessary to address this important issue. In addition, it has been shown that FASN increased expression directly correlates with clinical aggressiveness and poor prognosis in liver cancer.Citation33 Furthermore, FASN expression correlates with increased activated AKTCitation33 and loss of PTEN expressionCitation32 in human HCC. In mice, overexpression of an activated form of AKT or liver-specific deletion of PTEN leads to increased FASN expression and its mediated lipogenesis, resulting in hepatic steatosis.Citation33-35 In human HCC cells, it has been found that AKT/mTOR/RPS6 cascades promotes lipogenesis via multiple mechanisms, including increased mRNA expression of FASN and other lipogenic pathway genes, decreased FASN degradation, and augmented stability of SREBP1/2.Citation33 Altogether, these lines of evidence indicate that the AKT/mTOR pathway is the major regulator of lipogenesis along hepatocarcinogenesis.
Figure 3. Levels of FASN mRNA along human hepatocarcinogenesis. (A) Expression levels of FASN in normal livers (NL; n = 12), surrounding non-tumorous liver tissues (SL; n = 133) and corresponding HCC samples (n = 133) as assessed by real-time RT-PCR. (B) Comparison of FASN levels in surrounding non-tumorous livers associated with different etiologic factors for HCC development. (C) Comparison of FASN levels in HCC associated with different etiologic factors involved in hepatocarcinogenesis. Tukey–Kramer's test: ***P<0.0001. Abbreviations: ASH, alcoholic steatohepatitis; HBV, hepatitis B virus; HCV, hepatitis C virus; NASH, non-alcoholic steatohepatitis.
Functionally, FASN suppression has been demonstrated to be detrimental to HCC growth in vitro.Citation31,33,36 Specifically, silencing of FASN using siRNA or inhibition of FASN activity via the small molecule C75 led to decreased HCC cell proliferation and increased apoptosis. Mechanistically, it has been suggested that FASN may regulate the AKTCitation33 and p38MAPKCitation36 cascades. However, overexpression of FASN in the liver, either alone or in combination with other putative oncogenes, such as activated N-Ras or c-Met, does not trigger tumor formation in mice.Citation37 Consistently, liver-specific SREBP1C and SERPB1a transgenic mice showed an increased lipid biosynthesis paralleled by the upregulation of FASN, ACC and SCD1, leading to hepatic steatosis,Citation38 but no liver tumor development. Together, the results suggest that FASN and its mediated de novo lipogenesis are not oncogenic per se.
FASN is required for AKT and AKT/c-Met driven hepatocarcinogenesis in mice
While virtually all studies investigating whether FASN expression is required for liver tumor development have been performed in vitro, we sought to determine the requirement of FASN and its mediated de novo lipogenesis in hepatocarcinogenesis using genetic approaches. For this purpose, we applied conditional FASN KO miceCitation39 and various oncogene driven HCC mouse models, including AKT, AKT/c-Met and c-Met/β-catenin models.
Previous studies from our laboratory showed that hydrodynamic transfection of an activated form of AKT1 (myr-AKT) induces increased lipogenesis and, eventually, HCC formation over long-term.Citation33 To investigate whether FASN expression is required for myr-AKT driven liver tumor development, we hydrodynamically transfected myr-AKT and Cre recombinase (AKT/Cre mice) into FASNfl/fl mice.Citation37 Strikingly, while AKT overexpression in control mice led to malignant liver tumor formation by 22–28 weeks post-injection, in accordance with previous findings,Citation33 none of the AKT/Cre mice exhibited either liver enlargement or increased liver weight. Histologically, most of the liver parenchyma of AKT control mice was occupied by multiple hepatocellular tumors, whereas none of AKT/Cre mice displayed preneoplastic and neoplastic lesions. Similar results were obtained when we overexpressed myr-AKT in liver-specific FASN KO mice (AlbCre;FASNfl/fl mice). At the molecular level, we found that ablation of FASN in AKT/Cre mice resulted in decreased protein expression of Rictor (rapamycin-insensitive companion of mTOR), the main component of the mammalian target of rapamycin complex 2 (mTORC2), leading to the downregulation of phosphorylated/activated AKT.Citation37 The critical role of Rictor/mTORC2 in mediating activated AKT driven hepatocarcinogenesis was further validated using conditional Rictorfl/fl mice.Citation37 Specifically, ablation of Rictor in hepatocytes prevented myr-AKT driven hepatocarcinogenesis in mice, recapitulating the phenotypes observed in conditional FASN KO mice.Citation33 Altogether, these data indicate that FASN expression is indispensable for AKT-induced hepatocarcinogenesis, presumably due to its positive regulation over Rictor expression and mTORC2 activity.Citation37
Mounting evidence indicates that activation of the c-Met cascade is a key oncogenic event along human hepatocarcinogenesis.Citation17 c-Met encodes the receptor tyrosine kinase for hepatocyte growth factor (HGF). Our recent study has shown that co-expression of AKT and c-Met (AKT/c-Met) triggers rapid liver tumor development in mice, with all AKT/c-Met mice being required to be killed by 6 to 8 weeks' post-injection.Citation40 Importantly, we found that FASN is highly expressed in AKT/c-Met HCC cells.Citation40 To establish whether c-Met might confer resistance to FASN depletion along AKT hepatocarcinogenesis, we determined the effect of disrupting de novo lipogenesis in AKT/c-Met mice.Citation40 Therefore, we hydrodynamically transfected AKT, c-Met, and Cre plasmids into FASNfl/fl mice, thus allowing the simultaneous expression of AKT and c-Met oncogenes while deleting FASN in the same subset of mouse hepatocytes (AKT/c-Met/Cre). As a control, AKT, c-Met, and pT3-EF1α (empty vector) were co-injected into FASNfl/fl mice (AKT/c-Met/pT3). Of note, all AKT/c-Met/pT3 mice developed liver tumors by 8 weeks' post injection, whereas no macroscopic or histopathological alterations were detected in the livers of AKT/c-Met/Cre mice up to 15 weeks post hydrodynamic injection.Citation40 Similar results were obtained we expressed AKT/c-Met into liver-specific FASN KO mice (AlbCre;FASNfl/fl mice).Citation40 In summary, the present results indicate that c-Met confers aggressiveness and reduces tumor latency along AKT-driven hepatocarcinogenesis. Nonetheless, AKT/c-Met tumors remain fully dependent on FASN activity to develop.
FASN is dispensable for c-met/β-catenin induced liver cancer development
Previous studies indicate that not all HCC tumor samples show elevated FASN expression. Recently, we established another murine HCC model by hydrodynamically overexpressing c-Met and activated β-catenin in the mouse liver (c-Met/β-catenin).Citation41 Genomic studies have recently shown that this new murine model recapitulates a subset of human HCC.Citation41 Of note, we found that in contrast to that described in HCC induced by AKT or AKT/c-Met protooncogenes, the expression of FASN and other lipogenic pathway genes, such as ACC, is not elevated in c-Met/β-catenin tumor tissues (). Thus, we sought to determine whether FASN expression is required for c-Met/β-catenin driven hepatocarcinogenesis. For this purpose, 2 distinct approaches were applied. In the first approach, we hydrodynamically transfected c-Met and β-catenin together with Cre plasmids into FASNfl/fl mice (c-Met/β-catenin/Cre), thus allowing the simultaneous expression of c-Met and β-catenin oncogenes while deleting FASN in a subset of mouse hepatocytes (). As a control, c-Met, β-catenin, and pT3-EF1α (empty vector) were co-injected into FASNfl/fl mice (c-Met/β-catenin/pT3) (). Noticeably, we found that both c-Met/β-catenin/Cre and c-Met/β-catenin/pT3 injected FASNfl/fl mice developed lethal burden of HCC by ∼8 weeks post injection (). Histologically, well to moderately differentiated HCC were found in both cohorts of mice (). Importantly, immunostaining demonstrated the absent expression of FASN protein in c-Met/β-catenin/Cre HCC cells (), but tumor cells remained highly proliferative (). To complement this study, we hydrodynamically injected c-Met/β-catenin into liver-specific FASN KO mice (AlbCre;FASNfl/fl mice) as well as control littermates (FASNfl/fl mice) (). Once again, c-Met/β-catenin overexpression induced HCC formation in wild-type and liver-specific FASN KO mice with the same latency and histology (). Altogether, our study suggests that FASN expression is not upregulated in c-Met/β-catenin mouse HCC, and FASN is dispensable for hepatocarcinogenesis induced by c-Met and β-catenin oncogenes. The immunohistochemical patterns of FASN, ACC, and Ki-67 in wild-type (not injected or injected with empty plasmid), AKT, AKT/c-Met, and c-Met/β-catenin mice are summarized in .
Figure 4. Expression of FASN and ACC is not elevated in c-Met/β-catenin mouse tumor tissues (right panel), when compared with HCCs induced by AKT/c-Met overexpression (middle panel). Scale bar: 200µm. Abbreviations: WT, wild-type; FASN, fatty acid synthase; ACC, acetyl-CoA carboxylase; HE, hematoxylin and eosin staining.
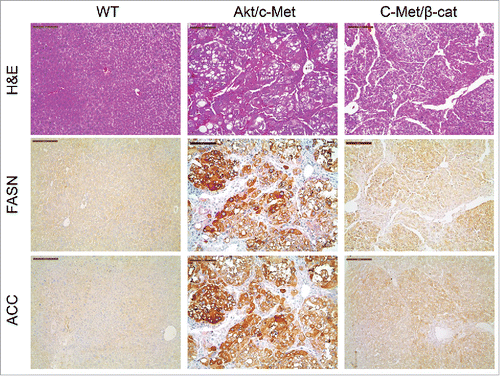
Figure 5. FASN expression is not upregulated along c-Met/β-catenin driven hepatocarcinogenesis. (A) Study design: activated forms of c-Met and β-catenin were hydrodynamically injected together with Cre into FASNfl/fl mice (c-Met/β-catenin/Cre, n = 4). This method allows the deletion of FASN while simultaneously expressing c-Met and β-catenin in the same FASNfl/fl hepatocytes. As a control, c-Met and β-catenin were injected in the same mice together with the empty vector (c-Met/β-catenin/pT3, n = 4). (B) Survival curve of c-Met/β-catenin/Cre mice (n = 4) and c-Met/β-catenin/pT3 injected FASNfl/fl mice (n = 4), P = 0.16. (C) Histologically, well to moderately differentiated HCC were found in both cohorts of mice and immunostaining demonstrated the low protein expression of FASN in c-Met/β-catenin/Cre HCC cells. Scale bar: 200µm for H&E and FASN, 100µm for Ki-67. Abbreviations: FASN, fatty acid synthase; HE, hematoxylin and eosin staining.
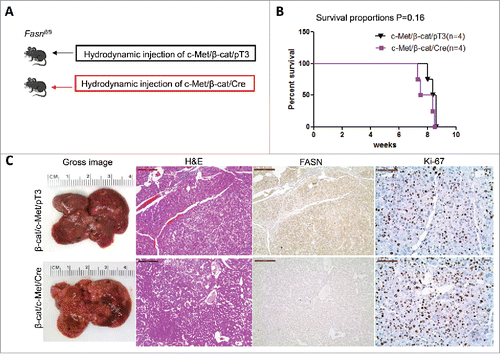
Figure 6. Genetic ablation of FASN in the mouse liver does not affect tumor development driven by c-Met/β-catenin co-expression. (A) Study design: Overexpression of c-Met/β-catenin promotes the development of multiple hepatocellular carcinomas both in FASNfl/fl mice with an intact FASN gene (n = 4) and in those where FASN was deleted by the Cre system (AlbCre;FASNfl/fl mice; n = 4). (B) c-Met/β-catenin induced HCC formation in wild-type and liver-specific FASN KO mice with the same latency and histology. (C) Survival curve of FASNfl/fl mice (n = 4) and liver-specific FASN KO mice (AlbCre;FASNfl/fl mice; n = 4) injected with c-Met/β-catenin, P = 0.35. Scale bar: 200µm for H&E and FASN, 100 µm for Ki-67. Abbreviations: FASN, fatty acid synthase; HE, hematoxylin and eosin staining.
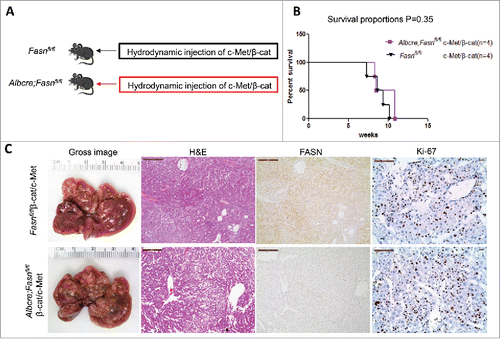
Table 1. Summary of the immunohistochemical patterns of FASN, ACC, and Ki-67 detected in wild-type livers (Wild-type), livers injected with empty vector (Empty vector) as well as in hepatocellular lesions (both preneoplastic and neoplastic) developed in AKT, AKT/c-Met, and c-Met/β-catenin mice.
It is important to underline that FASN(−) c-Met/β-catenin mouse HCCs are highly proliferative (). Clearly, the proliferating tumor cells require additional fatty acids, at the minimal, for cell membrane production during cell cycle progression. Ablation of FASN completely eliminates de novo lipogenesis pathway in tumor cells. Yet this does not affect c-Met/β-catenin tumor cell growth. Based on this body of evidence, it is plausible to hypothesize that c-Met/β-catenin tumor cells are most likely dependent on exogenous fatty acid uptake for their growth and proliferation. It has been suggested that lipids in circulating lipoprotein particles provide an exogenous source of fatty acids for tumor cells, although this process remains poorly understood.Citation42 This process requires very low-density lipoproteins (VLDL) or triglyceride-rich chylomicrons to be hydrolyzed by lipoprotein lipase (LPL). The free fatty acids can then be captured by cellular fatty acid transporters (CD36 and SLC27A family members) into the tumor cells.Citation43 In a recent study, we found that LPL is upregulated in human HCC samples; and high expression of LPL is associated with poor prognosis.Citation44 In human HCC cell lines, silencing FASN or LPL or culturing the cells in lipoprotein-deficient medium led to significantly decreased cell proliferation. In particular, when FASN and LPL were concomitantly silenced, the growth inhibition was significantly more pronounced than silencing of FASN or LPL alone.Citation44 These data strongly suggest that both de novo lipogenesis and exogenous fatty acid uptake may contribute to hepatocarcinogenesis. In c-Met/β-catenin HCC samples, we found that, indeed, LPL expression is elevated.Citation44 Conditional LPL Knockout mice have been generated.Citation45 However, they have not been used to study the requirement of LPL and its mediated exogenous fatty acid uptakes along tumorigenesis to date. Clearly, it would be of high importance to investigate whether LPL is required for c-Met/β-catenin driven hepatocarcinogenesis using conditional LPL knockout mice.
In summary, our genetic studies suggest the oncogene-dependent requirement of FASN and its mediated lipogenesis during hepatocarcinogenesis.
Targeting FASN and its mediated de novo lipogenesis for the treatment of HCC
Because increased de novo lipid biosynthesis is a frequent metabolic event in tumorigenesis, many efforts have been made to develop strategies targeting lipogenesis for cancer treatment.Citation21 As a major enzyme in lipogenesis, FASN has been the main target for developing small molecule inhibitors.Citation46,47 Virtually all these small molecules inhibit β-ketoacyle-reductase, which is critical for FASN's enzymatic activity. A complete list of FASN inhibitors can be found in a recent review.Citation21 Among them, C75 is an early developed FASN inhibitor and has been widely used as a pharmacological agent to evaluate FASN inhibitor for cancer treatment. For instance, it has been shown that C75 has anti-tumor activity in xenograft models of breast,Citation48 lung,Citation49 prostateCitation50 and renalCitation51 cancers, and it prevents breast cancer formation in HER2 transgenic mice.Citation52 TVB-2640 is the most recent orally available FASN inhibitor. It is currently in phase I clinical trial for the treatment of solid tumors (https://clinicaltrials.gov/ct2/show/NCT02223247).
In addition to FASN, other genes in the de novo lipogenic pathway have been pursued as drug targets as well. Among them, several molecules against ACC have been developed.Citation21 In a recent study, it was shown that ND-646, an allosteric ACC1/2 inhibitor, suppresses tumor growth in both xenograft and genetic engineered mouse models of non-small cell lung cancer, either alone or in combination with standard-of-care drug carboplatin,Citation53 supporting the further testing of ACC inhibitors for cancer treatment.
In HCC, currently, there are no studies assessing whether pharmacological inhibition of FASN and/or ACC can lead to tumor regression in preclinical models. Based on our studies, we would predict that FASN and/or ACC inhibitors are likely to have therapeutic potential against at least a subset of human HCCs. Importantly, valid biomarkers, such as immunostaining of FASN protein and ACC protein in HCC tumor samples, may be required to identify patients who may be most likely to benefit from anti-FASN/ACC based therapy. As increased FASN expression is tightly linked to the increased AKT/mTOR signaling, it is also possible to use p-AKT or p-RPS6 immunostaining as biomarkers for patient selection. Because Sorafenib is still the standard target therapy for advanced stage HCC, future studies are also required to evaluate whether anti-FASN/ACC drugs can be used in combination with Sorafenib for the treatment of HCC. Finally, it is important to consider the possible mechanisms of resistance to lipogenic pathway inhibitors. Most likely, based on our findings, HCC cells might compensate the loss of de novo lipogenesis by inducing exogenous fatty acid uptake. Thus, small molecules which target fatty acid transporters or LPL may be highly beneficial in combination with anti-FASN/ACC inhibitors for HCC treatment. Of note, some bile acids, such as ursodeoxycholic acid (UDCA), have been found to be efficient inhibitors of liver fatty acid transporter.Citation54 It would be of great interest to further test whether UDCA can be combined with FASN/ACC inhibitors for HCC treatment in preclinical models.
In summary, our and other studies have demonstrated the importance of FASN and its mediated de novo lipogenic metabolic cascade in hepatocarcinogenesis. It is of high importance to note that HCCs induced by different oncogenes are either addicted to or independent from de novo lipogenesis to grow. Future studies are required to evaluate the therapeutic potential of anti-lipogenic drugs for the treatment of HCCs, especially in those patients whose HCC shows elevated activation of the AKT/mTOR/lipogenesis cascade.
Materials and methods
Constructs
The plasmids used in the study, including pT3-EF1α-ΔN90-β-catenin, pT3-EF1α-c-Met, and pCMV-SB have been described previously.Citation55 All plasmids were purified using the Endotoxin-free Maxi Prep Kit before injecting into mice.
Hydrodynamic injection and mouse monitoring
FASNfl/fl mice as well as AlbCre;FASNfl/fl line in C57BL/6 background were described previously.Citation56 Hydrodynamic transfection was performed as described.Citation57 In brief, the plasmids encoding the gene(s) of interest along with sleeping beauty transposase (SB) in a ratio of 25:1 were diluted in 2 ml saline (0.9% NaCl), filtered through 0.22 μm filter, and injected into the lateral tail vein of the mice in 5 to 7 seconds. Mice were monitored and killed when they become moribund. Mice were housed, fed, and monitored in accordance with protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Quantitative reverse transcription real-time polymerase chain reaction (qRT-PCR)
Validated Gene Expression Assays for human FASN (ID: Hs01005622_m1) and β- Actin (ID: 4333762T) were purchased from Applied Biosystems (Foster City, CA). PCR reactions were performed with 100 ng of cDNA on the whole sample collection and cell lines, using an ABI Prism 7000 Sequence Detection System and TaqMan Universal PCR Master Mix (Applied Biosystems). Cycling conditions were: 10 min of denaturation at 95°C and 40 cycles at 95°C for 15 s and at 60°C for 1 min. Quantitative values were calculated by using the PE Biosystems Analysis software and expressed as N target (NT). NT = 2- ΔCt, wherein ΔCt value of each sample was calculated by subtracting the average Ct value of the target gene from the average Ct value of the β- Actin gene.
Immunohistochemical staining
Liver specimens were fixed in 4% paraformaldehyde and embedded in paraffin. For immunohistochemistry, deparaffinized sections were incubated in 3% H2O2 dissolved in 1X phosphate-buffered saline (PBS) for 30 minutes to quench the endogenous peroxidase. For antigen retrieval, slides were microwaved in 10 mM citrate buffer (pH 6.0) for 12 minutes. Subsequently, slides were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: anti-FASN (Cell Signaling Technology, #3180; 1:150), anti-ACC (Cell Signaling Technology, #3676; 1:100), and anti-Ki-67 (Thermo Fisher Scientific, #MA5–14520; 1:150). The immunoreactivity was visualized with the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), using Vector NovaRED™ (Vector Laboratories) as the chromogen. Slides were counterstained with Mayer's hematoxylin. The intensity of immunostaining was defined semi-quantitatively with a scale from “−“ to “+++” (−, negative; +, weak; ++, moderate; +++, strong). The immunohistochemical expression patterns of FASN and Ki-67 in the various mouse models are summarized in .
Statistical analysis
Data analysis was performed with Prism 6 (GraphPad, San Diego, CA). Kaplan-Meier analysis was used to compare the survival rate of 2 cohorts. Statistical differences among various groups were assessed using the Tukey–Kramer's test. All graphs represent mean ± SEM.
Abbreviations
| ACLY | = | adenosine triphosphate citrate lyase |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| ELOVL | = | elongation of very long chain fatty acids protein |
| FASN | = | fatty acid synthase |
| HCC | = | hepatocellular carcinoma |
| mTOR | = | mammalian target of rapamycin |
| mTORC | = | mTOR complex |
| Raptor | = | regulatory-associated protein of mTOR |
| Rictor | = | rapamycin-insensitive companion of mTOR |
| FA | = | fatty acid |
| PTEN | = | phosphatase and tensin homolog |
| RPS6 | = | ribosomal protein S6 |
| SREBP | = | sterol regulatory element binding protein |
| HGF | = | hepatocyte growth factor |
| SCD | = | stearoyl-CoA desaturase |
| UDCA | = | ursodeoxycholic acid |
| ACC | = | Acetyl-CoA carboxylase |
| LPL | = | lipoprotein lipase |
| VLDL | = | low-density lipoproteins |
| siRNA | = | small RNA interference |
| TKIs | = | kinase inhibitors |
| MAPK | = | mitogen-activated protein kinase |
| PI3K | = | phosphoinositide-3-kinase |
| MUSFA | = | monounsaturated fatty acids |
| PUSFA | = | polyunsaturated fatty acids |
| FADS | = | Fatty acid desaturase |
| CE | = | cholesterol ester |
| TG | = | triglyceride |
| PL | = | phospholipid |
| TCA | = | tricarboxylic acid |
| EGFR | = | epidermal growth factor receptor |
| N-Ras | = | neuroblastoma Ras viral oncogene homolog |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr. Clay F. Semenkovich (Washington University, St. Louis, MO) for providing FASNfl/fl mice.
Funding
This work was supported by a grant from the Italian Association Against Cancer (AIRC; grant number IG 12139) to DFC; grants NIH R01CA136606 and R21CA187306 to XC; P30DK026743 for UCSF Liver Center.
Related Research Data
References
- Pascual S, Herrera I, Irurzun J. New advances in hepatocellular carcinoma. World J Hepatol 2016; 8:421-38; PMID: 27028578; http://dx.doi.org/10.4254/wjh.v8.i9.421
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011; 365:1118-27; PMID: 21992124; http://dx.doi.org/10.1056/NEJMra1001683
- Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993; 328:1797-801; PMID: 7684822; http://dx.doi.org/10.1056/NEJM199306243282501
- Knudsen ES, Gopal P, Singal AG. The changing landscape of hepatocellular carcinoma: etiology, genetics, and therapy. American J Pathol 2014; 184:574-83; http://dx.doi.org/10.1016/j.ajpath.2013.10.028
- Choo SP, Tan WL, Goh BK, Tai WM, Zhu AX. Comparison of hepatocellular carcinoma in Eastern versus Western populations. Cancer 2016; [Epub ahead of print]; http://dx.doi.org/10.1002/cncr.30237
- Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010; 4(15 Suppl):14-22; http://dx.doi.org/10.1634/theoncologist.2010-S4-14
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378-90; PMID: 18650514; http://dx.doi.org/10.1056/NEJMoa0708857
- Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016; 150:835-53; PMID: 26795574; http://dx.doi.org/10.1053/j.gastro.2015.12.041
- Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology 2011; 140:1410-26; PMID: 21406195; http://dx.doi.org/10.1053/j.gastro.2011.03.006
- Kalyan A, Nimeiri H, Kulik L. Systemic therapy of hepatocellular carcinoma: current and promising. Clin Liver Dis 2015; 19:421-32; PMID: 25921671; http://dx.doi.org/10.1016/j.cld.2015.01.009
- Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008; 48:1312-27; PMID: 18821591; http://dx.doi.org/10.1002/hep.22506
- Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015; 149:1226-39 e4; PMID: 26099527; http://dx.doi.org/10.1053/j.gastro.2015.05.061
- Monga SP. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015; 148:1294-310; PMID: 25747274; http://dx.doi.org/10.1053/j.gastro.2015.02.056
- Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 2013; 23:1422-33.
- Delire B, Starkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Invest 2015; 45:609-23; PMID: 25832714; http://dx.doi.org/10.1111/eci.12441
- Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol 2014; 60:855-65; PMID: 24308993; http://dx.doi.org/10.1016/j.jhep.2013.11.031
- Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol 2014; 60:442-52; PMID: 24045150; http://dx.doi.org/10.1016/j.jhep.2013.09.009
- Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol 2014; 49:9-23; PMID: 24318021; http://dx.doi.org/10.1007/s00535-013-0907-x
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID: 21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013; 18:153-61; PMID: 23791484; http://dx.doi.org/10.1016/j.cmet.2013.05.017
- Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016; 16:732-49; PMID: 27658529; http://dx.doi.org/10.1038/nrc.2016.89
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7:763-77; PMID: 17882277; http://dx.doi.org/10.1038/nrc2222
- Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism 2014; 63:895-902; PMID: 24814684; http://dx.doi.org/10.1016/j.metabol.2014.04.003
- Herrera E, Amusquivar E. Lipid metabolism in the fetus and the newborn. Diabetes Metab Res Rev 2000; 16:202-10; PMID: 10867720; http://dx.doi.org/10.1002/1520-7560(200005/06)16:3%3c202::AID-DMRR116%3e3.0.CO;2-
- Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, Kunnumakkara AB. ATP citrate lyase (ACLY): a promising target for cancer prevention and treatment. Curr Drug Targets 2015; 16:156-63; PMID: 25537655; http://dx.doi.org/10.2174/1389450115666141224125117
- Peck B, Schulze A. Lipid desaturation - the next step in targeting lipogenesis in cancer? The FEBS J 2016; 283:2767-78; PMID: 26881388; http://dx.doi.org/10.1111/febs.13681
- Wang C, Ma J, Zhang N, Yang Q, Jin Y, Wang Y. The acetyl-CoA carboxylase enzyme: a target for cancer therapy? Expert Rev Anticancer Ther 2015; 15:667-76; http://dx.doi.org/10.1586/14737140.2015.1038246
- Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell Metab 2013; 18:465-9; PMID: 23973332; http://dx.doi.org/10.1016/j.cmet.2013.08.002
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID: 22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab 2012; 16:414-9; PMID: 23000402; http://dx.doi.org/10.1016/j.cmet.2012.09.002
- Hao Q, Li T, Zhang X, Gao P, Qiao P, Li S, Geng Z. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep 2014; 32:2471-6; PMID: 25231933
- Zhu X, Qin X, Fei M, Hou W, Greshock J, Bachman KE, Wooster R, Kang J, Qin CY. Combined phosphatase and tensin homolog (PTEN) loss and fatty acid synthase (FAS) overexpression worsens the prognosis of Chinese patients with hepatocellular carcinoma. Int J Mol Sci 2012; 13:9980-91; PMID: 22949843; http://dx.doi.org/10.3390/ijms13089980
- Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011; 140:1071-83; PMID: 21147110; http://dx.doi.org/10.1053/j.gastro.2010.12.006
- Ono H, Shimano H, Katagiri H, Yahagi N, Sakoda H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Viana AY, et al. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes 2003; 52:2905-13; PMID: 14633850; http://dx.doi.org/10.2337/diabetes.52.12.2905
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A 2004; 101:2082-7; PMID: 14769918; http://dx.doi.org/10.1073/pnas.0308617100
- Gao Y, Lin LP, Zhu CH, Chen Y, Hou YT, Ding J. Growth arrest induced by C75, A fatty acid synthase inhibitor, was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol Ther 2006; 5:978-85; PMID: 16855382; http://dx.doi.org/10.4161/cbt.5.8.2883
- Li L, Pilo GM, Li X, Cigliano A, Latte G, Che L, Joseph C, Mela M, Wang C, Jiang L, et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J Hepatol 2016; 64:333-41; PMID: 26476289; http://dx.doi.org/10.1016/j.jhep.2015.10.004
- Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. The J Biol Chem 1999; 274:30028-32; PMID: 10514488; http://dx.doi.org/10.1074/jbc.274.42.30028
- Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, Semenkovich CF. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab 2005; 1:309-22; PMID: 16054078; http://dx.doi.org/10.1016/j.cmet.2005.04.002
- Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, Li X, Latte G, Mela M, Evert M, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016; 6:20484.
- Tao J, Xu E, Zhao Y, Singh S, Li X, Couchy G, Chen X, Zucman-Rossi J, Chikina M, Monga SP. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology 2016; 64:1587-605; PMID: 27097116; http://dx.doi.org/10.1002/hep.28601
- Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 2013; 52:585-9; PMID: 24001676; http://dx.doi.org/10.1016/j.plipres.2013.08.005
- Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res 2009; (50 Suppl):S86-90; PMID: 19033209
- Cao D, Song X, Che L, Li X, Pilo MG, Vidili G, Porcu A, Solinas A, Cigliano A, Pes GM, et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int 2016; 37(1):80-89; PMID: 27264722; http://dx.doi.org/10.1111/liv.13183
- Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R, Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem 2004; 279:25050-7; PMID: 15028738; http://dx.doi.org/10.1074/jbc.M401028200
- Jones SF, Infante JR. Molecular Pathways: Fatty Acid Synthase. Clinical cancer research : an official journal of the American Association for Cancer Res 2015; 21:5434-8; http://dx.doi.org/10.1158/1078-0432.CCR-15-0126
- Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 2010; 6:551-62; PMID: 20373869; http://dx.doi.org/10.2217/fon.10.11
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res 2000; 60:213-8; PMID: 10667561
- Relat J, Blancafort A, Oliveras G, Cufi S, Haro D, Marrero PF, Puig T. Different fatty acid metabolism effects of (−)-epigallocatechin-3 gallate and C75 in adenocarcinoma lung cancer. BMC Cancer 2012; 12:280; PMID: 22769244; http://dx.doi.org/10.1186/1471-2407-12-280
- Chen HW, Chang YF, Chuang HY, Tai WT, Hwang JJ. Targeted therapy with fatty acid synthase inhibitors in a human prostate carcinoma LNCaP/tk-luc-bearing animal model. Prostate Cancer Prostatic Dis 2012; 15:260-4; PMID: 22565411; http://dx.doi.org/10.1038/pcan.2012.15
- Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J Urol 2008; 180:729-36; PMID: 18555493; http://dx.doi.org/10.1016/j.juro.2008.03.186
- Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene 2005; 24:39-46; PMID: 15489885; http://dx.doi.org/10.1038/sj.onc.1208174
- Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 2016; 22:1108-19; PMID: 27643638; http://dx.doi.org/10.1038/nm.4181
- Nie B, Park HM, Kazantzis M, Lin M, Henkin A, Ng S, Song S, Chen Y, Tran H, Lai R, et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology 2012; 56:1300-10; PMID: 22531947; http://dx.doi.org/10.1002/hep.25797
- Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, Kay MA, Wang R, Bishop JM. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007; 104:14771-6; PMID: 17785413; http://dx.doi.org/10.1073/pnas.0706578104
- Li L, Che L, Tharp KM, Park HM, Pilo MG, Cao D, Cigliano A, Latte G, Xu Z, Ribback S, et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016; 63:1900-13; PMID: 26910791; http://dx.doi.org/10.1002/hep.28508
- Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014; 184:912-23; PMID: 24480331; http://dx.doi.org/10.1016/j.ajpath.2013.12.002