
The molecular mechanisms charged with preventing tumor initiation and disease progression remain to be fully elucidated. Replicative senescence arising from telomere shortening was first described in 1961 by Hayflick and Moorehead (i.e., Hayflick limit) and has since been established to serve as a major suppressor of disease development in humans, including cancer.Citation1 Moreover, the anticancer activities of senescence programs can be induced by a variety of stimuli and cellular stressors, including DNA damage, oncogene activation, and oxidative stress. Likewise, whereas high dose chemotherapy typically induces apoptosis, low doses of these same agents often promote senescence, an event that enhances T cell-mediated immunosurveillance and cytolysis.Citation1 It should also be noted that senescent cells are readily detected in premalignant neoplasms, but not in their fully malignant counterparts, suggesting that (i) senescence programs suppress tumorigenesis, and (ii) escaping the grasp of senescence represents a key step in the progression of human neoplasms.Citation2 As such, it stands to reason that enhancing our understanding of how senescence is initiated and maintained may provide for novel strategies designed to inhibit tumorigenesis by reactivating senescence in developing and progressing carcinomas.
Senescence and its tumor suppressing functions are most frequently associated with the activation of 2 signaling nexuses, namely p16INK4A→Rb and p19ARF→p53.Citation1,3 Previous investigations by the Jackson Laboratory established that Oncostatin M (OSM)-mediated Stat3 activation could elicit senescence in p16- and p53-deficient human mammary epithelial cells (HMECs), and that this event could be circumvented by robust expression of c-Myc, leading to malignant transformation.Citation4 Similarly, aberrant c-Myc expression was also determined to be sufficient in overriding the senescent promoting activities of the TGF-β pathway in p16/p53-deficient HMECs engineered to express oncogenic Ras.Citation5 Based on the parallels between OSM- and TGF-β-mediated senescence in HMECs, Bryson et al Citation6 hypothesized that OSM may commandeer the TGF-β signaling system to elicit senescence in p16/p53-deficient HMECs. Accordingly, pharmacologic and genetic inactivation of the TGF-β signaling system prevented OSM from inducing senescence in HMECs, indicating a role for autocrine TGF-β signaling in coupling OSM to senescent programs. More importantly, OSM-mediated activation of Stat3 led to its physical interaction with Smad3, leading to their nuclear accumulation and induction of a TGF-β-responsive gene signature. Interestingly, c-Myc expression was repressed robustly by both OSM and TGF-β in HMECs, consistent with the role of c-Myc as a proto-oncogene and inhibitor of senescence. Indeed, engineering p16/p53-deficient HMECs to constitutively express c-Myc engendered their malignant transformation and ability to grow in an anchorage-independent manner, as well as elicited their acquisition of invasive and epithelial-mesenchymal transition (EMT) phenotypes in response to OSM and TGF-β. Moreover, following its inactivation of cellular senescence, aberrant c-Myc expression also imparted oncogenic activity upon OSM and enabled this cytokine to stimulate HMEC invasion and extracellular matrix remodeling in a manner reflective of those operant in supporting tumor development and metastatic progression. Collectively, these important findings highlight the cooperative functions that Stat3:Smad3 complexes play in stimulating senescence, and consequently, in inhibiting mammary tumorigenesis. Equally important, these findings also reinforce the central necessity of aberrant c-Myc activity to override senescence programs, and in doing so, convert OSM from a tumor suppressor to a tumor promoter ().
Figure 1. Mechanism of OSM-mediated senescence and its inactivation by c-Myc in premalignant mammary epithelial cells. Premalignant mammary epithelial cells that lack expression of p16 and p53 readily acquire senescent phenotypes in the presence of OSM. The ability of OSM to induce senescence requires autocrine TGF-β signaling and the formation of novel Stat3:Smad3 complexes (Circle 1) that accumulate in the nucleus to repress c-Myc expression, while simultaneously inducing that of p16 and p21 (Circle 2). Escaping senescence programs during malignant progression can be initiated by aberrant c-Myc expression, which endows OSM with tumor promoting functions and the ability to induce breast cancer cells to grow in an anchorage-independent manner, to exhibit increased invasiveness, and to undergo EMT programs driven by Snail (Circle 3). Red lettered circles correspond to provided unanswered questions and future directions (right panel). Abbreviations: AIG, anchorage-independent growth. IL-6, interleukin-6; IL-11, interleukin-11; LIF, leukemia inhibitory factor; MMPs, matrix metalloproteinases; OSMR, OSM receptor; TβR-I, TGF-β type I receptor; TβR-II, TGF-β type II receptor; TGF-βR, TGF-β receptors.
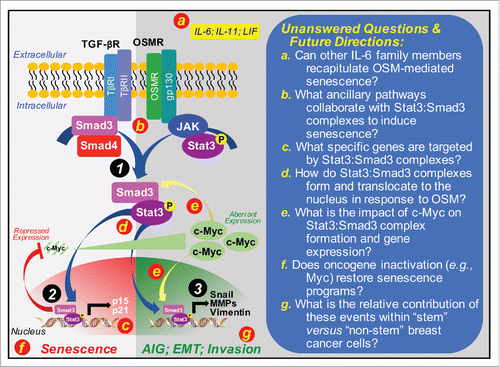
A provocative facet of this study relates to the dichotomous roles of OSM to both suppress and promote mammary tumorigenesis, an event governed by aberrant c-Myc expression. Although intriguing, the findings of this study raise several important unanswered questions related to OSM-mediated senescence (). For instance, as a member of the interleukin-6 (IL-6) family of cytokines, OSM shares the common transmembrane signaling receptor, gp130,Citation7 suggesting that cytokines belonging to this family hold the potential recapitulate OSM activity in breast cancers. Indeed, upregulated expression and activity of IL-6, IL-11, or leukemia inhibitory factor have all been associated with disease progression and poor prognoses in breast cancer patients. As such, future studies need to ascertain the role of other IL-6 family members in regulating senescence via the formation of Stat3:Smad3 complexes, as well as determine the relative contribution of Stat3-independent signaling systems in mediating the dichotomous actions of these cytokines, particularly in response to tumor inflammation and immune surveillance. Similar research efforts need to be directed at identifying the specific subsets of genes activated by Stat3:Smad3 complexes in senescent cells, and at determining the impact of aberrant c-Myc activity on the formation and function of Stat3:Smad3 complexes as premalignant cells escape senescent programs. Likewise, while it is clear that autocrine TGF-β signaling is essential for OSM-mediated senescence, it remains entirely unclear as to precisely how OSM drives the formation and nuclear translocation of Stat3:Smad3 complexes in the absence of Smad3 phosphorylation by TGF-β type I receptors. Along these lines, future studies also need to assess whether (i) the oncogenic activities of c-Myc directly or indirectly target Stat3:Smad3 complexes, and (ii) c-Myc inactivation restores senescent phenotypes, thereby suppressing disease progression and enhancing immunologic clearance of arrested tumors. Finally, given the important associations between EMT programs and the expansion of cancer stem cells, additional research efforts aimed at establishing the specificity of Stat3:Smad3 complexes in “stem” versus “non-stem” breast cancer cells is warranted. Ultimately, answering these intriguing questions will provide the necessary foundation to advance senescence-based therapies against developing and progressing human breast cancers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-496; PMID: 24954210; http://dx.doi.org/10.1038/nrm3823
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature 2005; 436:642; PMID: 16079833; http://dx.doi.org/10.1038/436642a
- Pare R, Yang T, Shin JS, Lee CS. The significance of the senescence pathway in breast cancer progression. J Clin Pathol 2013; 66:491-495; PMID: 23539738; http://dx.doi.org/10.1136/jclinpath-2012-201081
- Kan CE, Cipriano R, Jackson MW. c-MYC functions as a molecular switch to alter the response of human mammary epithelial cells to oncostatin M. Cancer Res 2011; 71:6930-6939; PMID: 21975934; http://dx.doi.org/10.1158/0008-5472.CAN-10-3860
- Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW. TGF-β signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci USA 2011; 108:8668-8673; PMID: 21555587; http://dx.doi.org/10.1073/pnas.1015022108
- Bryson BL, Junk DJ, Cipriano R, Jackson MW. STAT3-mediated SMAD3 activation underlies Oncostatin M-induced senescence. Cell Cycle 2016; 16(4):319-334; PMID: 27892764; http://dx.doi.org/10.1080/15384101.2016.1259037
- Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol 2014; 26:54-74; PMID: 24552665; http://dx.doi.org/10.1016/j.smim.2014.01.001