
ABSTRACT
Ubiquitination serves as a degradation mechanism of proteins, but is involved in additional cellular processes such as activation of NFκB inflammatory response and DNA damage repair. We highlight the E2 ubiquitin conjugating enzymes, E3 ubiquitin ligases and Deubiquitinases that support the metastasis of a plethora of cancers. E3 ubiquitin ligases also modulate pluripotent cancer stem cells attributed to chemotherapy resistance. We further describe mutations in E3 ubiquitin ligases that support tumor proliferation and adaptation to hypoxia. Thus, this review describes how tumors exploit members of the vast ubiquitin signaling pathways to support aberrant oncogenic signaling for survival and metastasis.
Ubiquitination signaling overview
Ubiquitin (Ub), a highly conserved 76-amino acid protein expressed in all cell types, has 7 lysine residues (K6, K11, K27, K29, K33, K48, K63) that can be polymerized into various linkages. The resulting linkage of Ub chains creates a certain topology that can be sampled by interacting proteins and dictates the fate of the substrate. For instance, K48- and K11-linked Ub chains adopt a “closed” or compact conformation and lead to 26S-mediated proteasomal degradation of substrates. In contrast, unanchored K63- or a mix of K63/M1-linked ubiquitination chains adopt an “open” conformation, and are involved in non-proteasomal functions such as TAK1 and IKK complex activation culminating in NFκB inflammatory signaling,Citation1,2 activation of DNA damage repair signalingCitation3 and B cell activation via MAPK by TAB2/TAB3.Citation4
Ubiquitination is a conserved multistep process that begins with the activation of ubiquitin with ATP by the E1 ubiquitin activating enzyme, followed by the formation of a thioester linkage between the ubiquitin transferred from the E1 to the cysteine in the active site of an E2 ubiquitin conjugating enzyme. The E3 ubiquitin ligase participates in the ubiquitination of a target substrate through the formation of an isopeptide bond between the carboxyl group of Gly76 of ubiquitin and the ϵ-amine of Lys in the substrate. Deubiquitinating enzymes (DUBs) remove the ubiquitin from the target substrates and recycle ubiquitin into the cytosolic pool.
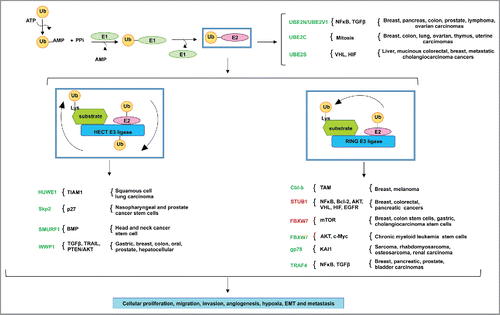
Ubiquitination regulates the function and signaling of a profusion of proteins in various cellular pathways. This review describes how various cancers take advantage of the misregulated expression of the members of the ubiquitination cascade for proliferation, survival and metastasis ().
Figure 1. The misregulated expression of E2 ubiquitin conjugating enzymes and E3 ubiquitin ligases in various human cancers. The ubiquitination reaction initiates with the activation of ubiquitin by ATP, in which ubiquitin is then transferred to the active site of E1 ubiquitin conjugating enzyme. The E1 transfers the ubiquitin to a Cys in the catalytic active site of the E2 ubiquitin conjugating enzyme. The HECT domain E3 ligases ubiquitinate the target substrates by 2 mechanisms: first, the ubiquitin is transferred from the active site of the E2 to the Cys in the active site of the E3, which then ubiquitinates the Lys residue in the target substrate. RING- and RING-related domain E3 ligases, in contrast, serve as scaffolds to ubiquitinate target substrates in one step: the E2 transfers the ubiquitin directly to the Lys residue in the target substrate. Various tumors take advantage of the misregulated expression of E2s and E3s for the aberrant activation of oncogenic pathways. E2s and E3s colored in green indicate the importance of their expression or overexpression in cancer, while those in red indicate their downregulated expression in cancer. These genomic events result in cancer cell proliferation, migration, invasion, angiogenesis, hypoxia, EMT and metastasis.
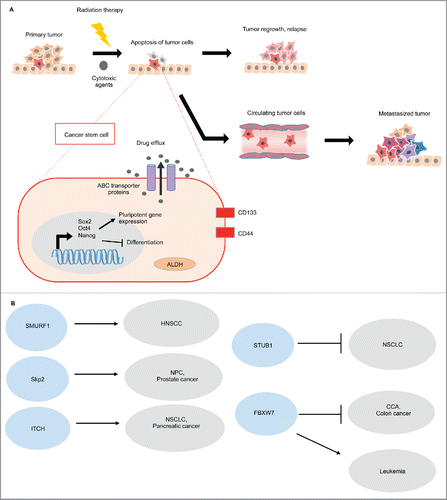
The challenging route to metastasis
Cancer progression eventually may lead to metastasis, which is the final stage responsible for more than 90% of all terminal cancer deaths. Various genomic abnormalities must be present to allow cells of a primary tumor to ignore apoptotic signals, proliferate and survive. Before metastasis occurs, tumor cells undergo phenotypic changes through epithelial-mesenchymal transition (EMT) similar to signaling events during embryonic development. EMT is marked by a loss of cell-cell adhesion through decreased expression of E-cadherin, increased motility by actin reorganization and upregulated expression of N-cadherin, Vimentin, Snail and Twist.Citation5 Cells are then able to move through the stroma, resist the episode of immune cells, survive and travel through the bloodstream. Finally, metastasized cells arrive at the secondary site, resisting rejection and apoptosis to form malignant microtumors. Eventually, these progress to clinically observable macro metastasized tumors. Only a small percentage of metastasized cells survive this migratory journey and successfully colonize the secondary tissue, classifying metastasis as a rare event.
Since most proteins undergo ubiquitination as a post-translational modification in most cell types, it is not surprising that cancer cells exploit the members of the ubiquitination pathway to stabilize aberrant oncogenic signaling. This review describes that the misregulated expression of E2s, E3 ligases and DUBs contributes to the signaling of various oncogenes, leading to cancer progression and metastasis.
The overexpression of E2s supports aberrant oncogenic signaling in tumor metastasis
E2s play an active role in the regulation of cell cycle progression, inflammation and the mechanisms by which they modulate cancer metastasis. There are approximately 40 E2 family members encoded by the human genome.Citation6 E2s share a highly conserved 150-amino acid catalytic core, the ubiquitin conjugating (UBC) domain, responsible for ubiquitination.
UBE2N/UBE2V1 modulates breast cancer metastasis
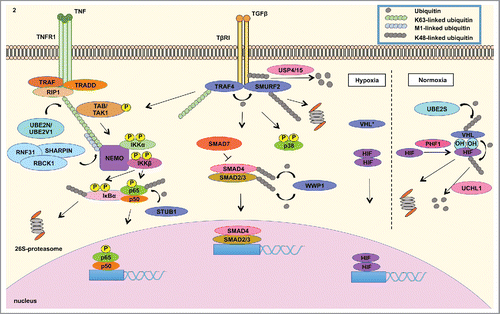
Ubiquitin-Conjugating Enzyme E2N (UBE2N, also known as UBC13), together with its co-factor UBE2V1 (also known as UEV1A), specifically builds Lys63-linked ubiquitin chains that are indispensable for NFκB inflammatory activation.Citation2 UBE2N is overexpressed in myriad tumors such as breast, pancreas, colon, prostate, lymphoma and ovarian carcinomas. UBE2N is required for breast cancer metastasis to the lung in vivo through TGFβ-mediated activation of TAK1 and p38, culminating in the expression of metastasis-associated genes CNN2, PLTP, IGFBP3, IL13RA2, CD44, VCAM-1 and ICAM-1 (). This requirement for UBE2N for metastatic spread is notable, given that UBE2N is not required for primary tumor formation.Citation7 ShRNA-mediated inhibition of UBE2N or treatment of breast cancer cells with SB203580, small molecule inhibitor of p38 MAPK, interestingly result in the suppression of breast cancer metastasis to the lung.Citation7
Figure 2. Misregulated expression of members of the ubiquitin cascade contributes to the aberrant signaling of various pathways in cancer. (LEFT) The UBE2N/UBE2V1 E2 ubiquitin conjugating enzyme complex catalyzes the Lys63-linked ubiquitination of NEMO that recruits the TAK1/TAB1/2 complex to activate the IKK complex, which is composed of IKKβ, IKKα and NEMO. IKKβ phosphorylates IκBα, which is Lys48-linked ubiquitinated and subsequently degraded by the 26S-proteasome.Citation2 This event releases NFκB to translocate into the nucleus to mediate the transcription of a signature of genes involved in inflammatory response. STUB1 E3 ligase negatively regulates NFκB signaling by catalyzing the degradation-inducing Lys48-linked ubiquitination of p65 subunit of NFκB.Citation23 (CENTER) In addition, cancer cells take advantage of overexpressed TRAF4 to modulate TGFβ signaling. TGFβ activation culminates in the nuclear translocation of SMAD2/3/4 complex to modulate gene transcription. SMAD7 is a negative regulator of TGFβ signaling by recruiting SMURF2 E3 ubiquitin ligase to ubiquitinate TβRI, leading to the proteasomal degradation of the receptor and mitigation of signalingCitation79 TGFβ signaling is regulated by TRAF4 E3 ligase mediated Lys48-linked ubiquitination of SMURF2 E3 ligase. The latter E3 ligase catalyzes the degradation signal of TGFβ receptor I (TβRI), in which these events mitigate the activation of the signaling pathway. TRAF4, on the other hand, is conjugated to Lys63-linked ubiquitin polymers to activate TAK1/TAB1/2 complex that induces the signaling of p38 MAPK and NFκB. TRAF4 further interacts with deubiquitinating enzyme USP15 and USP4, which remove the degradation signal from TGFβ receptor I. These events contribute to the stabilization of TGFβ signaling.Citation34 (RIGHT) Tumor adaptation to hypoxia is highly attributed to HIF signaling. Under normal oxygen level condition (normoxia), VHL E3 ligase binds to hydroxylated Proline residue in HIF catalyzed by PHD proteins. VHL then catalyzes the Lys48-linked ubiquitination of HIF, leading to proteasome degradation. UBE2S enzyme controls VHL protein stability. Under low oxygen conditions (hypoxia), inactivating of VHL (either by mutations or decreased expression) contributes to HIF isoform stabilization, which mediates the transcription of genes involved in tumor adaptation to hypoxia, including angiogenesis.Citation15,45 These signaling events governed by members of the ubiquitin cascade all contribute to EMT, cellular proliferation, migration, invasion, chemotherapy resistance and metastasis.
In addition, the cofactor for UBE2N, Ubiquitin-Conjugating Enzyme E2 Variant 1 (UBE2V1) is upregulated in breast cancer and increases the invasiveness and migration of breast cancer cells, including tumor growth and upregulated metastasis to lymph nodes and lung, an effect that is dependent upon functional UBE2N. These tumors exhibit increased expression of Matrix Metalloproteinase-1 (MMP1) through activation of NFκB signalingCitation8 (), in which MMPs are involved in extracellular matrix degradation for tumor cell migration and invasion.Citation9 ShRNA-mediated inhibition of UBE2V1 ablates breast tumor growth and metastasis in vivo. UBE2V1, in marked contrast to UBE2N, lacks the catalytic Cys and therefore catalytically inactive to perform the polyubiquitination of substrates.
It is not clear, however, whether these tumors exhibit the upregulated expression of both UBE2N and UBE2V1, or whether the upregulated expression of one member only is sufficient to aberrantly induce NFκB and TGFβ signaling to drive metastasis. The UBE2N/UBE2V1 complex, critical for NFκB signaling, is overexpressed in some breast cancer samples; this may contribute to the hyperactivation of an inflammatory response in the tumor microenvironment. Collectively, these studies suggest that UBE2N/UBE2V1, responsible for the formation of Lys63-linked ubiquitin chains which are important to activate signaling pathways as opposed to triggering protein degradation, represent potentially important targets for therapeutic intervention.Citation7-9
UBE2C regulates chromosomal alignment in mitosis
Overexpression of UBE2C (Ubiquitin-Conjugating Enzyme E2C, termed UBE2C or UBCH10) is detected in many cancers, including breast, colon, prostate, ovary, thymus, uterine and lung.Citation10-14 UBE2C functions with the Anaphase-promoting complex/cyclosome (APC/C) to ensure proper chromosome alignment and segregation in mitosis. UBE2C expression fluctuates during the cell cycle, peaking at prometaphase to regulate chromosomal segregation and decreasing in anaphase. Overexpression of UBE2C causes the missegregation of chromosomes (aneuploidy), chromosome misalignment and lagging, mitotic slippage and increased number of centrioles, in addition to a decrease in cyclin B1 levels. Mice engineered to overexpress UBE2C exhibit elevated lung tumor burden, including the emergence of lymphomas, lipomas, liver and skin tumorsCitation11 ().
As misregulated expression of UBE2C contributes to the emergence of tumors in tissues and cells from various lineages, it is possible that this E2 modulates chromosomal segregation during mitosis of different cancers, representing a target to treat a broad range of tumors.
UBE2S regulates E3 ligase VHL in hypoxia
UBE2S, or Ubiquitin-Conjugating Enzyme E2S (also known as E2-EPF ubiquitin carrier protein (UCP)), catalyzes the ubiquitination of von Hippel-Lindau (VHL) protein, targeting it for proteasomal degradation.Citation15 VHL mediates the stability of HIF transcription factors that induce expression of protumorigenic and mitogenic growth factor genes such as VEGF, MMPs, SNAIL, TWIST and PDGF involved in hypoxia, EMT, angiogenesis, migration, proliferation and metastasis ().Citation16
The overexpression of UBE2S and HIF1α, with low VHL expression, is detected in various tumors such as primary liver, mucinous colorectal and breast cancer, and in metastatic cholangiocarcinoma in soft tissue and metastatic colorectal cancer in lymph VHL controls the stability of hypoxia-inducible factors HIF-1 and HIF-2 that mediate the adaptation of cells to varying levels of oxygen. Overexpression of UBE2S in CAKI cells (clear cell carcinoma derived from metastatic skin site) and C8161 (highly invasive and metastatic human melanoma cells) contributes to increased degradation of VHL, while it increases HIF1α expression and VEGF transcription, contributing to the increased proliferation and metastasis to the lung.Citation15 UBE2S is also amplified in pancreatic and neuroendocrine prostate cancer.Citation17,18
In summary, this section illustrates that overexpression of E2 ubiquitin conjugating enzymes supports aberrant oncogenic signaling of inflammatory NFκB and TGFβ, receptor tyrosine kinases, mitogenic growth factors and HIF transcription factors. These E2s drive aneuploidy, proliferation, migration and metastasis of a variety of tumors.
The misregulated expression of E3 ubiquitin ligases in cancer
There are approximately 600 E3 ubiquitin ligases encoded by the human genome and the mechanism of ubiquitination of target substrates depends upon the conserved catalytic domains: RING (Really Interesting New Gene), HECT (Homology to E6AP C Terminus) and RING-related (PHD, LIM, F-box, B-box and U-box). RING and RING-related E3 ligases catalyze a one-step reaction of ubiquitin transfer from the E2 to the lysine residue in the substrate. In contrast, HECT E3 ligases catalyze a 2-step reaction: first the ubiquitin is transferred from the E2 to the cysteine in the active site of the E3 ligase, which then ubiquitinates the lysine residue in the target substrate.Citation19,20 ().
As with the E2 ubiquitin conjugating enzymes, misregulated expression of E3 ubiquitin ligases contributes to aberrant oncogenic signaling, metastasis and resistance to chemotherapy, including the modulation of pluripotency of cancer stem cells in tumor niches.
The downregulated expression of E3s in cancer
STUB1 – more than just an E3 ligase
Cancer cells take advantage of downregulated expression of STIP1 Homology And U-Box Containing Protein 1 (STUB1), also known as CHIP, a U-box ligase that functions as a chaperone for protein quality control and promotes the ubiquitination of various cell cycle regulators, such as c-Myc and SRC-3. STUB1 transcription is lower in malignant stage II and node-positive breast cancer than in stage I and node-negative patients. The downregulated expression of STUB1 upregulates NFκB signaling and anti-apoptotic proteins Bcl−2 and AKT, supporting inflammation, survival, invasiveness and metastatic potential of breast cancer cellsCitation21,22 In colorectal cancer, STUB1 is the E3 ubiquitin ligase that regulates the stability of p65 subunit of NFκB (). Downregulated STUB1 in colorectal cancer decreases the degradation of p65 subunit and increases the expression of NFκB-controlled VEGF, Cyclin D1, c-Myc, IL-8 and MMP-2 genes involved in angiogenesis and metastasis.Citation23
In pancreatic cancer, STUB1 is a tumor suppressor and it modulates the stability of EGFR via proteasomal-mediated degradation of this receptor tyrosine kinase (RTK). STUB1 regulates the phosphorylation of Tyr845 and Tyr1068 of EGFR, activating downstream PI3K/AKT and Src/FAK/paxillin signaling pathways. Downregulated STUB1 expression increases oncogenic EGFR signaling and sensitizes pancreatic cancer cells to RTK inhibitor, erlotinib, which leads to apoptosis and decreased tumor volume in vivo.Citation24
STUB1 modulates the proteasomal-mediated degradation of NFκB and EGFR oncogenes in various tumors (). Tumors harboring downregulated STUB1 expression exhibit aberrant NFκB signaling and some tumors may rely on RTK's for proliferative advantages. The role of STUB1 may extend beyond these pathways to include additional oncogenes in other tumors.
Despite its tumor suppressor function, enhanced STUB1 expression is observed in pancreatic, prostate breast and other cancers.Citation17,18,25 This suggests that further research will be required to unravel the tumor and context specific functions of this E3 ubiquitin-protein ligase.
FBXW7 – a key component of the SCF complex
F-Box And WD Repeat Domain Containing 7 (FBXW7) E3 ligase is a component of the SCF (SKP1, CUL-1, F-box protein) E3 ubiquitin ligase complex that regulates the stability of cell-cycle regulators such as c-Myc, cyclin E and Notch.Citation26
FBXW7 is downregulated in breast, colorectal, gastric and cholangiocarcinoma (CCA) tumors correlated with poor prognosis and survival, elevated tumor invasion and occurrence of metastasisCitation27-29 (). FBXW7 regulates the stability and turnover of mTOR. Downregulated FBXW7 expression increases mTOR levels that support the metastastic potential of CCA tumors to the liver and lung. These tumors are sensitive to mTOR inhibitor rapamycin, which impairs tumor growth.Citation29
In summary, this section illustrates that the downregulated expression of the E3 ubiquitin ligases stabilizes aberrant oncogenic signaling. Some of these reports, additionally, show that a rescue in the expression of these E3 ligases leads to increased degradation of key oncogenic signaling proteins, potentially resulting in the inhibition of tumor proliferation and metastasis.
The importance of the expression, or overexpression, of E3s in tumors
The contribution of Cbl-b to melanoma and breast cancer metastasis
The recruitment of immune system cells to the tumor microenvironment has been suggested to play an important role in tumor metastasis.Citation30 The E3 ligase activity of Casitas B-lineage lymphoma-b, Cbl-b or RNF56, is a negative regulator of anti-tumor function of natural killer (NK) cells. Genetic deletion of Cbl-b, or targeted inhibition of its E3 ubiquitin ligase activity, awakens the antitumor response of NK cells and educates these lymphoid cells to recognize and kill melanoma tumors, inhibiting lung metastasis. The TAM family of receptor tyrosine kinases, Tyro3, Axl and Mer, has been identified as substrates of Cbl-b for ubiquitination. Treatment of NK cells with selective TAM inhibitor, LDC1267, ablates melanoma and breast cancer metastasis.Citation31 This indicates that some E3 ligases, exemplified by Cbl-b, can modulate the immune system's ability to recognize and kill tumor cells.
HUWE1 controls cell-to-cell adhesion
In order for cancer cells to leave the primary tissue and metastasize to secondary sites, cell-to-cell adhesion must be disrupted for their movement through the stroma. The HECT, UBA, and WWE domain-containing protein 1 (HUWE1) E3 ubiquitin ligase has been implicated in the modulation of cell-to-cell adhesion. The expression of TIAM1, a guanine nucleotide exchange factor, at cell-to-cell junctions is critical to maintain cells in contact with one another. HUWE1-mediated degradation of TIAM1 leads to scattering, dissemination and invasion of epithelial cells, including the dissemination and local invasion of metastatic lung cells. Knockdown of HUWE1 decreases the dissemination of cells, leading to the stabilization of TIAM1 at cell-to-cell junctions. Stage I and stage II squamous cell lung carcinoma tissue samples show an inverse correlation between HUWE1 and TIAM1 expressionCitation32 (), suggesting that metastasis of lung cells may be modulated by the misregulated expression of HUWE1.
GP78 in sarcoma metastasis
The metastasis of sarcoma tumors is dependent upon the E3 ligase activity of GP78, also known as autocrine motility factor receptor (AMFR). GP78 is a RING-finger E3 ubiquitin ligase that localizes to the endoplasmic reticulum (ER) and it participates in ER-associated degradation (ERAD), a pathway that leads to the degradation of misfolded or denatured proteins. Inhibition of GP78 expression in highly metastatic human sarcoma cells inhibits lung metastasis, but it does not affect primary tumor growth. Stable gp78 knockdown decreases the survival of metastasized HT1080 sarcoma, RH30 rhabdomyosarcoma, HOS-MNNG osteosarcoma, including 786-O renal carcinoma (). The metastasis-suppressor KAI1 has been identified as a substrate of gp78-mediated degradation, in which an inverse correlation of KAI1 and GP78 expression is found in sarcoma samples. Downregulated gp78 leads to the accumulation of KAI1 that results in apoptosis and reduces the metastatic potential of sarcoma cells.Citation33 This event illustrates the importance of gp78 expression in sarcomas.
TRAF4 modulates inflammatory signaling of multiple tumors
Tumor necrosis factor receptor-associated factor 4 is a RING domain E3 ligase with well-documented signaling functions associated with the activation of TNFRs and IL-1R/TLRs, playing critical roles in immune system responses.Citation34
TRAF4 has been implicated in the regulation of both SMAD-dependent and SMAD-independent TGFβ receptor (TβRI)-induced signaling. TRAF4 ubiquitinates SMURF2 leading to the degradation of the latter E3 ligase and enhancement of TGFβ signaling. On the other hand, SMURF2 can ubiquitinate both TβRI and TRAF4 to terminate TGFβ signaling. TRAF4 also interacts with deubiquitinating enzyme USP15, which deubiquitinates TβRI upon SMURF2-mediated ubiquitination of TβRI, stabilizing this signaling pathway. In addition, TGFβ induces the Lys63-linked ubiquitination of TRAF4 to promote TAK1 activation, leading to p38 and NFκB signalingCitation34 ().
TRAF4 is amplified in invasive breast carcinoma, pancreatic adenocarcinoma, prostate cancer and bladder carcinoma.Citation18,25,35 (). TRAF4 overexpression contributes to poor overall survival in ovarian cancer and, in breast cancer, it is correlated with ERBB2 amplification and bone metastasis. TRAF4 further supports the expression of EMT makers such as N-cadherin, Vimentin, Fibronectin and TGFβ-associated expression of IL-11, PTHrP, CXCR4 and SNAIL. Knockdown of TRAF4 results in ablation of TGFβ-induced phosphorylation of SMAD2 and p38 MAPK, showing the importance of TRAF4 in the regulation of SMAD-dependent and -independent signalingCitation34
WWP1 – a phosphotyrosine binding E3
WW Domain Containing E3 Ubiquitin Protein Ligase 1 (WWP1) is a HECT domain E3 ubiquitin ligase that binds to phosphotyrosine (PPXY) domains in substrates.
WWP1 overexpression supports the proliferation and survival of oral, and hepatocellular carcinoma (HCC).Citation36,37 (). Additionally, WWP1 positively regulates PTEN/AKT signaling to support the proliferation and cell cycle progression of gastric tumors, contributing to poor survival and lymph node metastasisCitation38 In breast cancer, WWP1 is overexpressed in 51% of transformed cell lines and supports the expression of estrogen receptor and Insulin-like growth factor receptor-1, resulting in aberrant proliferation. Inhibition of WWP1 expression sensitizes TRAIL-resistant breast cancer cells to TRAIL-induced activation of the extrinsic apoptotic pathway. In addition, efficient inhibition of breast cancer proliferation has been achieved by combining knockdown of WWP1 with anti-estrogen tamoxifen drug therapy.Citation39-41 Lastly, WWP1 is frequently amplified and overexpressed in human prostate cancer, and is suggested to play a role in the inactivation of the TGFβ tumor suppressor pathway by inactivation of Smad2, Smad4 and TβR1 in human cancer.Citation42
Overall, this section illustrates that the overexpression of E3 ubiquitin ligases supports aberrant oncogenic signaling in various types of cancers. These reports show that inhibition of expression of each of these E3 ubiquitin ligase is sufficient to ablate tumor progression and metastasis.
Mutated E3 ligases proliferate different tumors
A list of mutations in E3 ubiquitin ligases, and their potential biologic effects in various tumors, is described in . We herein bring attention to the fact that mutated E3 ligases contribute to tumor hypoxia and vascularization, and upregulated inflammatory NFκB and growth factor mitogenic signaling pathways.
Table 1. Mutations in E3 ligases identified in cancers (partial list).
VHL adapts renal cell carcinoma to hypoxia
VHL is part of the VCB E3 ubiquitin ligase complex further composed of elongin B, elongin C and cullin-2. Under normoxia, VHL induces the ubiquitination of HIF1α for degradation. On the other hand, inactivation of VHL results in the stabilization of HIF isoforms that support vascularization and adaptation of tumors to hypoxiaCitation16 ().
Germ line mutations in tumor suppressor VHL cause von Hippel-Lindau syndrome characterized by the emergence of highly vascularized tumors, such as clear cell renal cell carcinoma (ccRCC), central nervous system hemagioblastoma, pheochromocytoma and pancreatic cysts.Citation16,43 Inactivated VHL is observed in approximately 80–90% of ccRCC, which includes >90% of allelic deletion or loss of heterozygosity, >50% mutations and approximately 10% of promoter hypermethylation.Citation44 Over 800 distinct mutations in VHL have been detected in sporadic and hereditary ccRCC, of which the majority results in loss of VHL function.Citation45
Mutated VHL causes tumors to become sensitive to PI3K p110β inhibitor, TGX221, which decreases the proliferation, migration and invasion of cells.Citation44 In silico analysis revealed that the majority of missense mutations affecting the surface of VHL impact the interaction of VHL with HIF isoforms, elongin B, elongin C and other binding partners such as p53 and PKC. For instance, the L101P mutation, and other VHL disease-causing mutations such as N78S, S80N and Y98H/N cause a dramatic loss-of-function in VHL and stabilize HIF1 isoforms. In addition, frameshift mutations in exons 1, 2 and 3, with the exception of Glu204fsX44 at the very end of exon 3, inactivate VHL and lead to increased stability of HIF isoforms (). Such alterations in VHL possibly affect the hydroxylation of HIF isoforms by prolyl 4-hydroxylases ().Citation45 Hence, the stabilization of HIF isoforms by inactivation of the E3 ligase activity of VHL possibly contributes to hypoxia and the highly vascularized characteristics observed in these cancers.
c-Cbl drives myeloproliferative neoplasms
Casitas B-Lineage Lymphoma, c-Cbl or RNF55, is member of the Cbl family of E3 ubiquitin ligases previously mentioned. Mutations in c-Cbl in myeloproliferative neoplasms usually occur due to acquired 11q uniparental disomy, an event similar to loss of heterozygosity in which a part of the DNA is lost from one chromosome but the remaining homolog is duplicated resulting in 2 copies at the locus per cell. Myeloproliferative neoplasms include chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML; BCR-ABL negative), myelofribrosis (MF), secondary acute myelogenous leukemia (sAML), acute myeloid leukemia and juvenile myelomonocytic leukemia (JMML).Citation46-49 Homozygous mutations in c-Cbl in myeloproliferative neoplasms contribute to poor survival rates of patients. Mutations in c-Cbl contribute to earlier presentation of JMML disease (age of 12 months at diagnosis) compared with patients without these alterations (29 months).Citation47,49
c-Cbl mutations themselves are able to lead to neoplasms, as they have been identified in JMML tumors that do not harbor any other mutated RAS, PTN11 or NF1 genes known to be involved in JMML.Citation47 The mutations S376F (aCML), H398Y (CMML), P417A (aCML) and R420Q (AML, aCML, MF, sAML) in c-Cbl when co-transfected with FLT3 confer IL3-independence in 32D cells reflecting their oncogenic transforming activities (). These mutations also impair c-Cbl-mediated ubiquitination of FLT3 and internalization of EGFR and PDGFR,Citation46,48,50 while they induce activated phospho-STAT5 signaling.Citation50 Mutation at the “hot spot” Y371 site of c-Cbl is rarely found in other myeloid neoplasms. The Y371C/D/H mutations located in the linker domain adjacent to the RING domain have been identified in JMML patients (). A similar phosphomimic mutation to Asp, Y371E, constitutively activates the autoubiquitination E3 ligase activity of c-Cbl and it interacts with EGFR,Citation51 showing that c-Cbl modulates RTK signaling.
Activating mutations in c-Cbl lead to constitutive RTK signaling by impairing receptor internalization and degradation, driving myeloproliferative disorders.
Mutations in RNF31 in ABC DLBCL
The Linear Polyubiquitin Chain Assembly Complex (LUBAC) is composed of RNF31 (or HOIP), RBCK1 (or HOIL-1) and SHARPIN. LUBAC catalyzes the linear ubiquitination of NEMO, a scaffold member of the IKK complex that activates the canonical NFκB signaling pathway ().
Activated B cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) malignant progression is dependent upon constitutive B-cell receptor (BCR) activation that upregulates NFκB signaling. Inhibition of RNF31 or SHARPIN decreases LUBAC-mediated linear polyubiquitination of NEMO and impaires IKK and NFκB activation, leading to a decrease in viability of ABC DLBCL.Citation52 In addition, inhibition of RNF31 sensitizes ABC DLBCL tumors to Bruton agammaglobulinemia tyrosine kinase (BTK) inhibitor, Ibrutinib, and IRF4 transcription factor inhibitor, Lenalidomine, leading to decreased viability of this subclass of B-cell lymphoma.
Two rare germline SNP polymorphisms in RNF31 are enriched 8-fold in ABC DLBCL biopsies (). These mutations, Q584H and Q622L, are found in the ubiquitin-associated domain of RNF31 known to interact with the ubiquitin-like domain of RBCK1, leading to upregulation of LUBAC-mediated linear polyubiquitination of NEMO, increased IKK and NFκB activation in ABC DLBCL tumors.Citation52
Inactivating driver mutations in FBXW7
Mutations in FBXW7 have been identified in various tumors (). The F-box domain of FBXW7 is important for the interaction with the SCF complex, while the WD40 domain recognizes phosphorylation sites in a conserved Cdc4 phospho-degron motif in the target substrate.Citation26 Inactivating mutations in the WD40 domain of FBXW7 often impair substrate binding and subsequently diminish protein turnover, contributing to aberrant oncogenic signaling of targets mentioned below.
T cell acute lymphoblastic leukemia (T-ALL) is a malignant neoplasm characterized by mutations mostly in NOTCH1 and FBXW7.Citation53 Mutations in FBXW7, studied in a population of pediatric Chinese T-ALL patients, contribute to poor overall survival and higher incidence of tumor relapse. These FBXW7 mutations affect codons R465, R479, R505 and R689 in the WD40 domain, critical for the interaction with the PEST domain of NOTCH1. These mutations may further impact signaling pathways beyond NOTCH1, since FBXW7 mediates the stability of oncogenic proteins such as mTOR, Cyclin E and MYC.
In addition, mutations in FBXW7 are frequently found in various cancers, but particularly uterine, colorectal, bladder and cervical carcinoma.Citation54-57 In melanoma, some FBXW7 mutations have been reported in the absence of the classic BRAF V600E and NRAS G12D, G13R and Q61K/L/S mutations. This may indicate that mutations in FBXW7 are drivers of cancer progression.Citation58 As a proof-of-concept for the inactivating mutations in FBXW7, knockdown of FBXW7 in melanoma cell lines leads to the accumulation of NOTCH1, HEY1 and downstream effectors Cyclin E, Aurora A and MYC, resulting in increased tumorigenesis.Citation59-61 In addition, this event leads to the upregulation of angiogenesis-promoting genes such as IL-6, CXCL2, SERPINE1 and PGF1, and increased phosphorylation of STAT3. Mice grafted with melanoma cells with knockdown FBXW7, treated with NOTCH1 inhibitors dibenzazepine (DBZ) and compound E, showed substantial melanoma tumor shrinkage.Citation58
Other inactivating mutations in FBXW7 were identified in various advanced cancers such as colorectal, squamous head and neck, bladder, cervix, endometrial, liver, ovarian, mesothelioma, pancreatic and teratoma. In these tumors, most of these mutations concomitantly occur with mutations in TP53, followed by KRAS, PI3KCA and APC.Citation62 As mTOR is a downstream target of FBXW7, tumors harboring inactivating mutations in FBXW7 are sensitive to mTOR inhibitors. For instance, treatment of patients, whose tumors exhibit mutated FBXW7, with mTOR inhibitors (Sirolimus, Everolimus, and Temsirolimus) resulted in tumor shrinkage.Citation62 In addition, Temsirolimus showed promising results in a patient with metastatic lung adenocarcinoma with mutated FBXW7 R465H, leading to shrinkage of mediastinal lymphadenopathy ().Citation63
E3 ubiquitin ligases in pluripotent cancer stem cells
Conventional chemotherapies often fail to target metastasized tumors, which may be due to the emergence of a subpopulation of cells, the cancer initiating cells or cancer stem cells (CSCs). CSCs are not only capable of self-renewal and differentiation like normal stem cells, but they are also capable of tumor initiation, relapse, chemotherapeutic resistance and metastasis.
CSCs are characterized by upregulated expression of embryonic stem cells markers, SOX2 (SRY-Box 2), OCT4 (Octamer-Binding Protein 4) and NANOG (Nanog Homeobox). These transcription factors are the key regulators of pluripotency, activating self-renewal-associated genes and inhibiting differentiation by suppressing lineage-specific transcription factors. Expression of these regulators confers stem cell-like characteristics, and silencing them results in decreased tumorigenicity. CSC's express surface markers CD133 and CD44, including increased expression of aldehyde dehydrogenase (ALDH) and increased ATP binding cassette (ABC) transporter drug efflux, which all contribute to chemotherapy resistanceCitation64 ().
Figure 3. E3 ubiquitin ligases in pluripotent stem cells. (A) Cancer stem cells are characterized by the upregulation of Sox2, Oct4, and Nanog, which activates self-renewal associated genes and inhibits cellular differentiation. Cell surface markers, CD133 and CD44 are associated with CSC properties. Conventional chemotherapeutic agents target differentiated cells, thus quiescent CSCs are innately chemo-resistant. Moreover, CSCs show increased ABC multi-drug transporters, which pump the cytotoxic drugs out of the cells. CSCs also show high aldehyde dehydrogenase activity (ALDH), which detoxifies the aldehydes generated by the chemotherapeutic agents. As a result, the surviving CSCs can re-populate or metastasize, and these cancer cells that possess self-renewal advantages are very challenging to eradicate by using conventional chemotherapeutic agents.Citation64 (B) E3 ligases can either promote or suppress CSCs in different cancers.
E3 ligases are strongly associated with either promoting or suppressing the CSC population in various cancers (). Investigating the mechanisms by which E3 ligases confer stem-cell like characteristics to a subpopulation of tumorigenic cells is of great interest to develop novel compounds to selectively target CSC's, possibly overcoming metastasis.
SMURF1 in head and neck cancer stem cells
SMURF1 (SMAD specific E3 ubiquitin protein ligase 1) suppresses bone marrow morphogenic (BMP) signaling contributing to the maintenance of the CSC subpopulation (ALDHhigh/CD44high) in head and neck squamous cell carcinoma (HNSCC). BMP proteins are growth factor members of the TGFβ superfamily that restrict haematopoietic stem cell proliferation and induce cellular differentiation. The overexpression of SMURF1 in CSCs derived from HNSCC leads to decreased levels of SMADs 1/5/8 and attenuates BMP signaling, keeping cells in an undifferentiated stem-cell like phenotype.Citation65
Skp2 in nasopharyngeal and prostate cancer stem cells
Skp2 (S-phase kinase-associated protein 2 also known as FBXL1) is an F-box protein; one of its main targets for degradation is the G1/S cyclin-dependent kinase inhibitor p27, a tumor suppressor.
Overexpression of Skp2 correlates with poor prognosis of nasopharyngeal carcinoma (NPC), one of the most common head and neck carcinomas,Citation66 and of prostate cancerCitation67 (). NPC patients with high expression of Skp2 have a correlation with tumor recurrence and metastasis. Knockdown of Skp2 decreases sphere colony formation of NPC cell lines, and impairs the proliferation of the ALDH1+ subpopulation of cells, indicating reduced CSC phenotypes.Citation66 In prostate cancer, a small molecule inhibitor of Skp2 reduces prostate CSC population through p53-independent cellular senescence and inhibition of aerobic glycolysis. Moreover, the Skp2 inhibitor sensitizes prostate CSC's to doxorubicin and cyclophosphamide drug treatments.Citation67
Taken together, Skp2 promotes CSC properties and decreases drug treatment efficacy, showing the importance of targeting Skp2 to ablate CSC in tumor niches.
ITCH in lung cancer stem cells
ITCH is a HECT E3 ligase that performs essential regulatory functions in immune cells such as ubiquitination of Bcl10, PKC and PLC-γ, leading to nuclear translocation of NFκB and NFAT (nuclear factor of activated T-cells) during T-cell signaling activation. ITCH further regulates the stability of p63 and Notch.Citation68
Desmethylclomipramine (DCMI), identified via high-throughput screening as a specific inhibitor of E3 ligase activity of ITCH,Citation69 has shown promising chemotherapeutic action against non-small cell lung CSCs in patient samples whom acquired resistance to chemotherapeutic drugs Cisplatin, Gemcitabine and Paclitaxel. DCMI leads to reduced sphere forming ability and inhibition of proliferation of this subpopulation of lung cancer stem cells. In addition, shRNA-mediated silencing of ITCH decreases ALDH-positive lung CSCs and sensitizes cells to Gemcitabine-induced apoptosis.Citation70
FBXW7 in colonic and leukemia cancer stem cell maintenance
FBXW7 is known to target oncoproteins such as mTOR, cyclin-E, Jun, Myc and Notch1 for degradation, which are usually involved in the signaling of various cancers. The role of FBXW7 in the maintenance of normal stem cells has been previously shown.Citation71 However, the role of FBXW7 in the maintenance of CSCs can vary, depending on the type of tumor ().
In colon cancer, downregulated FBXW7 expression promotes EMT as shown by the increased epxression of mesenchymal stem cell markers, SOX2, OCT4 and NANOG, leading to invasive phenotype, greater tumor initiating potential and non-adherent growth ability. As previously mentioned, mTOR is a downstream target of FBXW7. Treatment of colon cancer cells with the mTOR inhibitor rapamycin suppresses their migration and tumor-sphere formation.Citation72 Loss of FBXW7 is also associated with stem-like competence in cholangiocarcinoma (CCA), and rapamycin treatment of CCA cells suppresses invasion, metastatic potential and tumor sphere formation.Citation29
In contrast, expression of FBXW7 is critical in the maintenance of leukemia-initiating cells (LICs) in chronic myeloid leukemia (CML), a disease associated with the BCR-ABL fusion protein. FBXW7 supports stem cell properties in LICs and maintains these cells in a quiescent state. Inhibition of FBXW7 expression decreases colony formation potential, eliminates leukemic cell infiltration in peripheral blood, spleen, liver and lungs, and leads to induction of apoptosis in a p53-dependent manner. In addition, knockdown of FBXW7 causes LIC's to differentiate and enter the cell cycle becoming sensitized to imatinib and cytosine arabinoside drug treatments.Citation73
In summary, the levels in the expression of E3 ubiquitin ligases maintain a subpopulation of cancer stem cells in an undifferentiated state (). Most of these reports show that inhibition of E3 ligase expression, with the exception of FBXW7 in LIC's, sensitizes CSC's to chemotherapeutic agents suggesting a clinical approach to eradicate this subpopulation of cells attributed to chemotherapy resistance and metastasis.
The misregulated expression of DUBs in metastatic cancers
Deubiquitinating enzymes (DUBs) play the reverse role of E3 ligases by cleaving the isopeptide bond between the ubiquitin and the substrate. DUBs have been described to play important roles in cellular processes, such as gene transcription, DNA repair and cell cycle progression. There are about 80 functional DUBs and they can be divided into 6 classes: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian-tumor proteases (OTUs), Machado-Joseph disease protein domain proteases (MJD), JAMM/MPN domain-associated metallopeptidases (JAMMs) and monocyte chemotactic protein-induced protein (MCPIP).Citation74
In cancer, DUBs can stabilize proteins by removing the degradation-inducing ubiquitin signal, contributing to aberrant signaling (). Many studies have shown that deregulation of DUBs is involved in cancer progression, recurrence and metastasis ().
Figure 4. Proposed model of the mechanism by which the misregulated expression of E2s, E3s and DUB's may contribute to tumorigenesis and metastasis. The information from the reports described in this review reveals that inhibition of E2s, E3s and DUB's with either small interfering RNA or small molecule inhibitors is sufficient to inhibit the progression and metastasis of tumors. However, some of these reports show that the misregulated expression of the members of the ubiquitination pathway occurs in multiple tumors. It is not clear whether a subpopulation of cells within a tumor, for instance, may rely on the misregulated expression of one member only or on a combination of the misregulated expression of these proteins to support aberrant oncogenic signaling. In addition, it is not clearly demonstrated whether the misregulated expression of E2s and E3s stabilizes oncogenic signaling via the catalysis of non-proteasomal ubiquitin linkages, while DUB's remove ubiquitin linkages that would otherwise lead to proteasomal degradation. Nevertheless, we describe a common conclusion from multiple reports that members of the ubiquitination pathway drive tumor aggressiveness and culminate in metastasis.
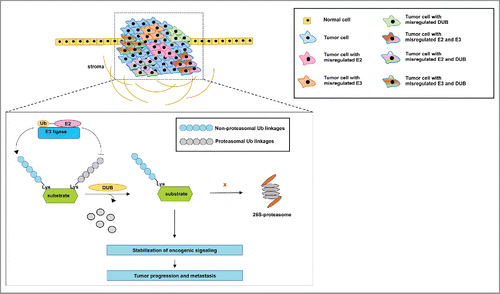
Table 2. Aberrant expression of DUBs associated with cancers.
USP7 promotes APL, aggressive prostate cancer, and NSCLC metastasis
USP7 (also known as HAUSP, Herpesvirus-associated ubiquitin protease) plays a crucial role in regulating tumor suppressors p53 and PTEN. A small molecule inhibitor of USP7 (HBX 41,108) inhibits USP7-mediated deubiquitination of p53 and induces apoptosis in colon carcinoma, suggesting the therapeutic potential in targeting USP7.Citation75 USP7 is frequently overexpressed in non-small cell lung carcinoma (NSCLC) and also correlates with lymph node metastasis. Inhibition of USP7 upregulates E-cadherin and downregulates Vimentin and N-cadherin, indicating USP7 promotes EMT in NSCLC.Citation76
Moreover, USP7 may contribute to cancer by modulating the nuclear localization of PTEN. Acute promyelocytic leukemia (APL) harboring the t(15; 17) chromosomal translocation, which encodes the PML–RARα fusion protein that inhibits differentiation, exhibits aberrant nuclear exclusion of PTEN. As USP7 co-localizes to nuclear bodies, studies using prostate and colon tumor models have shown USP7 to interact with and deubiquitinate PTEN, leading to the nuclear exclusion and impairment of tumor suppressor function of PTEN. In addition, USP7 is overexpressed in prostate cancer tissue, in which the USP7/PTEN signaling axis is misregulated.Citation77
Interestingly, murine ES cells expressing mutant p53 can retain pluripotency and normal karyotype through a mechanism in which USP7, Nedd4, Aurora kinase, Trim24 and the chaperonin containing TCP1 complex (CCT) participate in binding mutant p53, thus inducing a conformational shift toward a WT conformation. Exploiting this gain-of-function mechanism has been proposed as a possible path toward future p53-targeted cancer therapy.Citation78
USP4 and USP9X support oncogenic signaling in various tumors
Ubiquitin-specific peptidase 4 (USP4) is overexpressed in multiple cancers, such as breast and lung cancers (). Some cancers rely on aberrant TGFβ signaling for the purposes of EMT, proliferation, invasion and metastasis. As previously mentioned, TGFβ-induced activation of TβRI eventually results in the ubiquitination of TβRI and subsequent membrane internalization and degradation of the receptor, terminating downstream signaling. USP4 overexpression is detected in invasive breast carcinoma and it enhances aberrant TGFβ signaling. A mechanism by which this oncogenic signaling occurs has been proposed. AKT-mediated phosphorylation of USP4 at a consensus motif stabilizes this DUB, which in turn deubiquitinates the TGFβ-induced activated TβRI and leads to the stabilization of downstream activation of phosphorylated SMAD2 and SMAD2/SMAD4 complex formation (). USP4-mediated stabilization of TGFβ signaling leads to the expression of metastasis-inducing genes such as IL-11, CXCR4 and MMPs in breast cancer cells.Citation79
In addition, the overexpression of another DUB - USP9X - has been detected in follicular lymphoma, breast and colon adenocarcinomas, and small cell lung carcinomaCitation80 (). Various lymphomas, such as B- and mantle-cell lymphomas and multiple myeloma usually rely on the overexpressed MCL1, a member of the pro-survival BCL family of proteins. In multiple myeloma, overexpression of USP9X is correlated with poor survival. A mechanism by which MCL1 is stabilized has been proposed. The overexpression of USP9X results in the deubiquitination of MCL1 by this DUB, stabilizing pro-survival oncogenic signaling in cancers. Although knocking down USP9X does not affect cell proliferation in vitro, it does sensitize tumors to ABT-737 small molecule antagonist of BCL proteins resulting in apoptosis.Citation80 More recently, in non-small cell lung carcinoma, inhibition of USP9X by either siRNA knockdown or via a small molecule inhibitor WP1130 decreased MCL1 expression and sensitized cells to radiation therapy.Citation81
UCH-L1 plays a contradicting role in different tumors
Overexpression of UCH-L1 (Ubiquitin C-terminal hydrolase L1) in gastric cancer promotes colony formation, migration and liver metastasis of gastric cancer cells in vitro due to significant upregulation of AKT and Erk1/2 signaling.Citation82 Moreover, UCH-L1 is overexpressed in highly invasive NSCLC and melanoma. In lung cells, UCH-L1 supports the EGF-induced activation of Akt, JNK, and p38 MAPK signaling.Citation83
In addition, high expression of UCH-L1 positively correlates with that of HIF1α and hence activation of hypoxia conditions in some breast and lung cancer, and it contributes to poor overall survival and distant metastasis-free survival in breast, lung and melanoma cancer. UCH-L1 is shown to deubiquitinate and stabilize HIF1α, inducing hypoxia (). UCHL1 expression supports breast and melanoma metastasis to lungs; inhibition of metastasis is achieved by a UCH-L1 inhibitor, LDN57444.Citation84
However, the role of UCH-L1 is not as straightforward as suggested by these studies. Another study reported that UCHL1 can be downregulated in other breast cancer tissue samples and cell lines due to frequent gene methylation. Overexpression of UCHL1 in MB231 breast cells significantly decreases proliferation due to G0/G1 cell cycle arrest and apoptosis.Citation85 Similarly, this DUB is downregulated in some prostate cancer samples and in LNCaP cell line, and overexpression of UCHL1 suppresses the proliferation and anchorage-independent growth of LNCaP cells.Citation86 Further research will be required to unravel the contradicting role of UCH-L1 in different cancers.
OTUB1 contributes to drug resistance and promotes metastasis
OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) plays diverse roles such as stabilizing p53, inhibiting K63-linked polyubiquitination for DNA double strand break repair by targeting UBE2N for degradation, the E2 enzyme that catalyzes this ubiquitin linkage in proteins members of the NFκB pathway. High expression of OTUB1 is detected in approximately 50% of colorectal cancer and correlates with lymph node status, liver, pelvic and ovary distant metastasis, in addition to low overall and progression-free survival rates. OTUB1 is also found to be important in prostate cancer by regulating androgen signaling.Citation87,88 In breast cancer, OTUB1 decreases the Lys48-linked ubiquitination of FOXM1 stabilizing the expression of the protein. FOXM1 is a transcription factor that is important for DNA damage responses and its overexpression is associated with genotoxic drug resistance in tumors. Hence, the overexpression of OTUB1 contributes to the aberrant proliferation and epirubicin resistance of MCF7 cells and correlates with poor survival and chemotherapy resistance of breast cancer cohort.Citation89 In summary, it is evident that some DUBS are either upregulated or downregulated in some tumors, promoting oncogenic signaling.
Concluding remarks
The information presented herein reveals a common pattern: the misregulated expression of various E2 ubiquitin conjugating enzymes, E3 ubiquitin ligases and DUBs supports aberrant oncogenic signaling in a plethora of tumors. Since the misregulated expression of these proteins overlaps among many different types of cancers, there may exist subpopulations of cells within tumors exhibiting the misregulated expression of a specific member of the ubiquitination pathway. Alternatively, cells may take advantage of a combinatorial misregulated expression of these proteins to support oncogenic signaling. Lastly, it is not clear whether E2s and E3s catalyze non-proteasomal ubiquitin linkages that stabilize the function of oncogenes, and/or DUB's remove the ubiquitin linkages that would otherwise result in the degradation of oncogenes. Several models are presented by which members of the ubiquitination pathway support oncogenic signaling in cancers (). Nevertheless, it is clear that the inhibition of a specific E2, E3 or DUB seems to halt tumor progression and metastasis.
The development of chemotherapeutic agents against E2s, E3s and DUBs, is an attractive strategy that may lead to the inhibition of multiple oncogenic pathways in tumors. So far, the FDA has approved very few drugs targeting members of the ubiquitination cascade. Proteasome inhibitors, Bortezomib and Carfilzomib, are limited to multiple myeloma and mantle cell lymphoma treatments. Various strategies to develop inhibitors to target E1s, E2s, E3s have been recently described.Citation90 For instance, targeting the MDM2 E3 ligase - a p53 negative regulator - with RG7112 (cis-imidazoline analogs) has been tested in clinical trials for the treatment of advanced solid and hematological malignancies, including liposarcoma.Citation91
An interesting concept to enhance the ubiquitination and degradation of “undruggable” oncogenic proteins is the development of protein-targeting chimeric molecules (PROTACs). These molecules are bifunctional in which they include an E3 ligase-recruiting moiety linked by a short linker to a ligand that targets the substrate of interest, placing them in proximity for ubiquitination leading to subsequent degradation.Citation90 Therefore, targeting the E3 ligases and their co-expressed substrates with PROTAC may be a promising therapeutic intervention in various tumors herein described.
Major advances have been made to target oncogenic protein kinases in clinical settings through the development of TKIs. Here, we present evidence from multiple papers demonstrating that mechanistic studies of the ubiquitination pathway will open new therapeutic approaches, potentially allowing the development of novel inhibitors to suppress cancer progression and metastasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank all laboratory members for advice and encouragement, particularly April N. Meyer.
Funding
We also acknowledge generous support from the UC 820 San Diego Foundation.
Related Research Data
References
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A 2013; 110:15247-52; PMID:23986494; http://dx.doi.org/10.1073/pnas.1314715110
- Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol 2013; 25:4-12; PMID:23312890; http://dx.doi.org/10.1016/j.coi.2012.12.005
- Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol 2009; 187:319-26; PMID:19948475; http://dx.doi.org/10.1083/jcb.200908074
- Ori D, Kato H, Sanjo H, Tartey S, Mino T, Akira S, Takeuchi O. Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. J Immunol 2013; 190:4037-45; PMID:23509369; http://dx.doi.org/10.4049/jimmunol.1300173
- Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016; 352:167-9; PMID:27124449; http://dx.doi.org/10.1126/science.aaf6546
- Clague MJ, Heride C, Urbe S. The demographics of the ubiquitin system. Trends Cell Biol 2015; 25:417-26; PMID:25906909; http://dx.doi.org/10.1016/j.tcb.2015.03.002
- Wu X, Zhang W, Font-Burgada J, Palmer T, Hamil AS, Biswas SK, Poidinger M, Borcherding N, Xie Q, Ellies LG, et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc Natl Acad Sci U S A 2014; 111:13870-5; PMID:25189770; http://dx.doi.org/10.1073/pnas.1414358111
- Wu Z, Shen S, Zhang Z, Zhang W, Xiao W. Ubiquitin-conjugating enzyme complex Uev1A-Ubc13 promotes breast cancer metastasis through nuclear factor-small ka, CyrillicB mediated matrix metalloproteinase-1 gene regulation. Breast Cancer Res 2014; 16:R75; PMID:25022892; http://dx.doi.org/10.1186/bcr3692
- Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 2006; 25:9-34; PMID:16680569; http://dx.doi.org/10.1007/s10555-006-7886-9
- Fujita T, Ikeda H, Kawasaki K, Taira N, Ogasawara Y, Nakagawara A, Doihara H. Clinicopathological relevance of UbcH10 in breast cancer. Cancer Sci 2009; 100:238-48; PMID:19038004; http://dx.doi.org/10.1111/j.1349-7006.2008.01026.x
- van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol 2010; 188:83-100; PMID:20065091; http://dx.doi.org/10.1083/jcb.200906147
- Narayan G, Bourdon V, Chaganti S, Arias-Pulido H, Nandula SV, Rao PH, Gissmann L, Durst M, Schneider A, Pothuri B, et al. Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: identification of candidate amplified and overexpressed genes. Genes Chromosomes Cancer 2007; 46:373-84; PMID:17243165; http://dx.doi.org/10.1002/gcc.20418
- Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, Hoang A, Efstathiou E, Sircar K, Navone NM, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res 2012; 18:666-77; PMID:22156612; http://dx.doi.org/10.1158/1078-0432.CCR-11-1867
- Takahashi Y, Ishii Y, Nishida Y, Ikarashi M, Nagata T, Nakamura T, Yamamori S, Asai S. Detection of aberrations of ubiquitin-conjugating enzyme E2C gene (UBE2C) in advanced colon cancer with liver metastases by DNA microarray and two-color FISH. Cancer Genet Cytogenet 2006; 168:30-5; PMID:16772118; http://dx.doi.org/10.1016/j.cancergencyto.2005.12.011
- Jung CR, Hwang KS, Yoo J, Cho WK, Kim JM, Kim WH, Im DS. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med 2006; 12:809-16; PMID:16819549; http://dx.doi.org/10.1038/nm1440
- Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science 2016; 352:175-80; PMID:27124451; http://dx.doi.org/10.1126/science.aaf4405
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015; 6:6744; PMID:25855536; http://dx.doi.org/10.1038/ncomms7744
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016; 22:298-305; PMID:26855148; http://dx.doi.org/10.1038/nm.4045
- Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 2014; 21:301-7; PMID:24699078; http://dx.doi.org/10.1038/nsmb.2780
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem 2009; 78:399-434; PMID:19489725; http://dx.doi.org/10.1146/annurev.biochem.78.101807.093809
- Jang KW, Lee KH, Kim SH, Jin T, Choi EY, Jeon HJ, Kim E, Han YS, Chung JH. Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-kappaB inactivation to regulate breast cancer cell invasion. J Cell Biochem 2011; 112:3612-20; PMID:21793045; http://dx.doi.org/10.1002/jcb.23292
- Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I, Yamaguchi Y, Ohie SH, Kobayashi Y, Seino Y, et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol 2009; 11:312-9; PMID:19198599; http://dx.doi.org/10.1038/ncb1839
- Wang Y, Ren F, Wang Y, Feng Y, Wang D, Jia B, Qiu Y, Wang S, Yu J, Sung JJ, et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappaB-mediated signaling in colorectal cancer. Carcinogenesis 2014; 35:983-91; PMID:24302614; http://dx.doi.org/10.1093/carcin/bgt393
- Wang T, Yang J, Xu J, Li J, Cao Z, Zhou L, You L, Shu H, Lu Z, Li H, et al. CHIP is a novel tumor suppressor in pancreatic cancer through targeting EGFR. Oncotarget 2014; 5:1969-86; PMID:24722501; http://dx.doi.org/10.18632/oncotarget.1890
- Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S, Mar C, Wan A, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015; 518:422-6; PMID:25470049; http://dx.doi.org/10.1038/nature13952
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83-93; PMID:18094723; http://dx.doi.org/10.1038/nrc2290
- Ibusuki M, Yamamoto Y, Shinriki S, Ando Y, Iwase H. Reduced expression of ubiquitin ligase FBXW7 mRNA is associated with poor prognosis in breast cancer patients. Cancer Sci 2011; 102:439-45; PMID:21134077; http://dx.doi.org/10.1111/j.1349-7006.2010.01801.x
- Iwatsuki M, Mimori K, Ishii H, Yokobori T, Takatsuno Y, Sato T, Toh H, Onoyama I, Nakayama KI, Baba H, et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. Int J Cancer 2010; 126:1828-37; PMID:19739118
- Yang H, Lu X, Liu Z, Chen L, Xu Y, Wang Y, Wei G, Chen Y. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015; 6:6310-25; PMID:25749036; http://dx.doi.org/10.18632/oncotarget.3355
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/10.1038/nm.3394
- Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014; 507:508-12; PMID:24553136; http://dx.doi.org/10.1038/nature12998
- Vaughan L, Tan CT, Chapman A, Nonaka D, Mack NA, Smith D, Booton R, Hurlstone AF, Malliri A. HUWE1 ubiquitylates and degrades the RAC activator TIAM1 promoting cell-cell adhesion disassembly, migration, and invasion. Cell reports 2015; 10:88-102; PMID:25543140; http://dx.doi.org/10.1016/j.celrep.2014.12.012
- Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B, Veenstra T, Hewitt SM, Helman LJ, Khanna C, et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med 2007; 13:1504-9; PMID:18037895; http://dx.doi.org/10.1038/nm1686
- Zhang L, Zhou F, Garcia de Vinuesa A, de Kruijf EM, Mesker WE, Hui L, Drabsch Y, Li Y, Bauer A, Rousseau A, et al. TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol Cell 2013; 51:559-72; PMID:23973329; http://dx.doi.org/10.1016/j.molcel.2013.07.014
- Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, Piscuoglio S, Wen HC, Lim RS, Shen R, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol 2015; 237:179-89; PMID:26095796; http://dx.doi.org/10.1002/path.4573
- Lin JH, Hsieh SC, Chen JN, Tsai MH, Chang CC. WWP1 gene is a potential molecular target of human oral cancer. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:221-31; PMID:23849376; http://dx.doi.org/10.1016/j.oooo.2013.05.006
- Cheng Q, Cao X, Yuan F, Li G, Tong T. Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma carcinoma cells through the activation of caspase3 and p53. Biochem Biophys Res Commun 2014; 448:248-54; PMID:24792179; http://dx.doi.org/10.1016/j.bbrc.2014.04.117
- Zhang L, Wu Z, Ma Z, Liu H, Wu Y, Zhang Q. WWP1 as a potential tumor oncogene regulates PTEN-Akt signaling pathway in human gastric carcinoma. Tumour Biol 2015; 36:787-98; PMID:25293520; http://dx.doi.org/10.1007/s13277-014-2696-0
- Chen C, Zhou Z, Ross JS, Zhou W, Dong JT. The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer 2007; 121:80-7; PMID:17330240; http://dx.doi.org/10.1002/ijc.22653
- Chen C, Zhou Z, Sheehan CE, Slodkowska E, Sheehan CB, Boguniewicz A, Ross JS. Overexpression of WWP1 is associated with the estrogen receptor and insulin-like growth factor receptor 1 in breast carcinoma. Int J Cancer 2009; 124:2829-36; PMID:19267401; http://dx.doi.org/10.1002/ijc.24266
- Zhou Z, Liu R, Chen C. The WWP1 ubiquitin E3 ligase increases TRAIL resistance in breast cancer. Int J Cancer 2012; 130:1504-10; PMID:21480222; http://dx.doi.org/10.1002/ijc.26122
- Chen C, Sun X, Guo P, Dong XY, Sethi P, Zhou W, Zhou Z, Petros J, Frierson HF, Jr., Vessella RL, et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007; 26:2386-94; PMID:17016436; http://dx.doi.org/10.1038/sj.onc.1210021
- Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011; 19:617-23; PMID:21386872; http://dx.doi.org/10.1038/ejhg.2010.175
- Feng C, Sun Y, Ding G, Wu Z, Jiang H, Wang L, Ding Q, Wen H. PI3Kbeta inhibitor TGX221 selectively inhibits renal cell carcinoma cells with both VHL and SETD2 mutations and links multiple pathways. Scientific reports 2015; 5:9465; PMID:25853938; http://dx.doi.org/10.1038/srep09465
- Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P, Moch H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Cancer Res 2011; 71:5500-11; PMID:21715564; http://dx.doi.org/10.1158/0008-5472.CAN-11-0757
- Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O'Keefe C, Hsi E, Paquette RL, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 2009; 27:6109-16; PMID:19901108; http://dx.doi.org/10.1200/JCO.2009.23.7503
- Muramatsu H, Makishima H, Jankowska AM, Cazzolli H, O'Keefe C, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 2010; 115:1969-75; PMID:20008299; http://dx.doi.org/10.1182/blood-2009-06-226340
- Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009; 113:6182-92; PMID:19387008; http://dx.doi.org/10.1182/blood-2008-12-194548
- Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009; 114:1859-63; PMID:19571318; http://dx.doi.org/10.1182/blood-2009-01-198416
- Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, Thiessen C, Tickenbrock L, Schwable J, Brandts C, et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 2007; 110:1004-12; PMID:17446348; http://dx.doi.org/10.1182/blood-2007-01-066076
- Kassenbrock CK, Anderson SM. Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J Biol Chem 2004; 279:28017-27; PMID:15117950; http://dx.doi.org/10.1074/jbc.M404114200
- Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, Wright GW, Zhao H, Yang Y, Xu W, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Dis 2014; 4:480-93; PMID:24491438; http://dx.doi.org/10.1158/2159-8290.CD-13-0915
- Yuan L, Lu L, Yang Y, Sun H, Chen X, Huang Y, Wang X, Zou L, Bao L. Genetic mutational profiling analysis of T cell acute lymphoblastic leukemia reveal mutant FBXW7 as a prognostic indicator for inferior survival. Ann Hematol 2015; 94:1817-28; PMID:26341754; http://dx.doi.org/10.1007/s00277-015-2474-0
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014; 507:315-22; PMID:24476821; http://dx.doi.org/10.1038/nature12965
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. Recurrent R-spondin fusions in colon cancer. Nature 2012; 488:660-4; PMID:22895193; http://dx.doi.org/10.1038/nature11282
- Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 2014; 5:5006; PMID:25233892; http://dx.doi.org/10.1038/ncomms6006
- Muller E, Brault B, Holmes A, Legros A, Jeannot E, Campitelli M, Rousselin A, Goardon N, Frebourg T, Krieger S, et al. Genetic profiles of cervical tumors by high-throughput sequencing for personalized medical care. Cancer Med 2015; 4:1484-93; PMID:26155992; http://dx.doi.org/10.1002/cam4.492
- Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, Brunner G, Cordon-Cardo C, Osman I, Rabadan R, et al. FBXW7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst 2014; 106:dju107; PMID:24838835; http://dx.doi.org/10.1093/jnci/dju107
- Sato M, Rodriguez-Barrueco R, Yu J, Do C, Silva JM, Gautier J. MYC is a critical target of FBXW7. Oncotarget 2015; 6:3292-305; PMID:25669969; http://dx.doi.org/10.18632/oncotarget.3203
- Fujii Y, Yada M, Nishiyama M, Kamura T, Takahashi H, Tsunematsu R, Susaki E, Nakagawa T, Matsumoto A, Nakayama KI. Fbxw7 contributes to tumor suppression by targeting multiple proteins for ubiquitin-dependent degradation. Cancer Sci 2006; 97:729-36; PMID:16863506; http://dx.doi.org/10.1111/j.1349-7006.2006.00239.x
- Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, Bayliss R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci U S A 2016; 113:13726-31; PMID:27837025; http://dx.doi.org/10.1073/pnas.1610626113
- Jardim DL, Wheler JJ, Hess K, Tsimberidou AM, Zinner R, Janku F, Subbiah V, Naing A, Piha-Paul SA, Westin SN, et al. FBXW7 mutations in patients with advanced cancers: clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS One 2014; 9:e89388; PMID:24586741; http://dx.doi.org/10.1371/journal.pone.0089388
- Villaruz LC, Socinski MA. Temsirolimus therapy in a patient with lung adenocarcinoma harboring an FBXW7 mutation. Lung cancer (Amsterdam, Netherlands) 2014; 83:300-1; PMID:24360397; http://dx.doi.org/10.1016/j.lungcan.2013.11.018
- Morrison BJ, Morris JC, Steel JC. Lung cancer-initiating cells: a novel target for cancer therapy. Target Oncol 2013; 8:159-72; PMID:23314952; http://dx.doi.org/10.1007/s11523-012-0247-4
- Khammanivong A, Gopalakrishnan R, Dickerson EB. SMURF1 silencing diminishes a CD44-high cancer stem cell-like population in head and neck squamous cell carcinoma. Mol Cancer 2014; 13:260; PMID:25471937; http://dx.doi.org/10.1186/1476-4598-13-260
- Wang J, Huang Y, Guan Z, Zhang JL, Su HK, Zhang W, Yue CF, Yan M, Guan S, Liu QQ. E3-ligase Skp2 predicts poor prognosis and maintains cancer stem cell pool in nasopharyngeal carcinoma. Oncotarget 2014; 5:5591-601; PMID:25015320; http://dx.doi.org/10.18632/oncotarget.2149
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013; 154:556-68; PMID:23911321; http://dx.doi.org/10.1016/j.cell.2013.06.048
- Aki D, Zhang W, Liu YC. The E3 ligase Itch in immune regulation and beyond. Immunol Rev 2015; 266:6-26; PMID:26085204; http://dx.doi.org/10.1111/imr.12301
- Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, Cavasotto CN, Knight RA, Ciechanover A, et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis 2014; 5:e1203; http://dx.doi.org/10.1038/cddis.2014.113
- Bongiorno-Borbone L, Giacobbe A, Compagnone M, Eramo A, De Maria R, Peschiaroli A, Melino G. Anti-tumoral effect of desmethylclomipramine in lung cancer stem cells. Oncotarget 2015; 6:16926-38; PMID:26219257; http://dx.doi.org/10.18632/oncotarget.4700
- Takeishi S, Nakayama KI. Role of Fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br J Cancer 2014; 111:1054-9; PMID:24853181; http://dx.doi.org/10.1038/bjc.2014.259
- Wang Y, Liu Y, Lu J, Zhang P, Wang Y, Xu Y, Wang Z, Mao JH, Wei G. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun 2013; 434:352-6; PMID:23558291; http://dx.doi.org/10.1016/j.bbrc.2013.03.077
- Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell 2013; 23:347-61; PMID:23518349; http://dx.doi.org/10.1016/j.ccr.2013.01.026
- D'Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther 2015; 147:32-54; PMID:25444757; http://dx.doi.org/10.1016/j.pharmthera.2014.11.002
- Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 2009; 8:2286-95; PMID:19671755; http://dx.doi.org/10.1158/1535-7163.MCT-09-0097
- Zhao GY, Lin ZW, Lu CL, Gu J, Yuan YF, Xu FK, Liu RH, Ge D, Ding JY. USP7 overexpression predicts a poor prognosis in lung squamous cell carcinoma and large cell carcinoma. Tumour Biol 2015; 36:1721-9; PMID:25519684; http://dx.doi.org/10.1007/s13277-014-2773-4
- Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008; 455:813-7; PMID:18716620; http://dx.doi.org/10.1038/nature07290
- Rivlin N, Katz S, Doody M, Sheffer M, Horesh S, Molchadsky A, Koifman G, Shetzer Y, Goldfinger N, Rotter V, et al. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc Natl Acad Sci U S A 2014; 111:7006-11; PMID:24778235; http://dx.doi.org/10.1073/pnas.1320428111
- Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX, et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-beta type I receptor. Nat Cell Biol 2012; 14:717-26; PMID:22706160; http://dx.doi.org/10.1038/ncb2522
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham-Anderson J, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010; 463:103-7; PMID:20023629; http://dx.doi.org/10.1038/nature08646
- Kushwaha D, O'Leary C, Cron KR, Deraska P, Zhu K, D'Andrea AD, Kozono D. USP9X inhibition promotes radiation-induced apoptosis in non-small cell lung cancer cells expressing mid-to-high MCL1. Cancer Biol Ther 2015; 16:392-401; PMID:25692226; http://dx.doi.org/10.1080/15384047.2014.1002358
- Gu YY, Yang M, Zhao M, Luo Q, Yang L, Peng H, Wang J, Huang SK, Zheng ZX, Yuan XH, et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumour Biol 2015; 36:8379-87; PMID:26018507; http://dx.doi.org/10.1007/s13277-015-3566-0
- Kim HJ, Kim YM, Lim S, Nam YK, Jeong J, Kim HJ, Lee KJ. Ubiquitin C-terminal hydrolase-L1 is a key regulator of tumor cell invasion and metastasis. Oncogene 2009; 28:117-27; PMID:18820707; http://dx.doi.org/10.1038/onc.2008.364
- Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, Kobayashi M, Hirota K, Itasaka S, Yoshimura M, et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat Commun 2015; 6:6153; PMID:25615526; http://dx.doi.org/10.1038/ncomms7153
- Xiang T, Li L, Yin X, Yuan C, Tan C, Su X, Xiong L, Putti TC, Oberst M, Kelly K, et al. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS One 2012; 7:e29783; PMID:22279545; http://dx.doi.org/10.1371/journal.pone.0029783
- Ummanni R, Jost E, Braig M, Lohmann F, Mundt F, Barett C, Schlomm T, Sauter G, Senff T, Bokemeyer C, et al. Ubiquitin carboxyl-terminal hydrolase 1 (UCHL1) is a potential tumour suppressor in prostate cancer and is frequently silenced by promoter methylation. Mol Cancer 2011; 10:129; PMID:21999842; http://dx.doi.org/10.1186/1476-4598-10-129
- Iglesias-Gato D, Chuan YC, Jiang N, Svensson C, Bao J, Paul I, Egevad L, Kessler BM, Wikstrom P, Niu Y, et al. OTUB1 de-ubiquitinating enzyme promotes prostate cancer cell invasion in vitro and tumorigenesis in vivo. Mol Cancer 2015; 14:8; PMID:25623341; http://dx.doi.org/10.1186/s12943-014-0280-2
- Zhou Y, Wu J, Fu X, Du W, Zhou L, Meng X, Yu H, Lin J, Ye W, Liu J, et al. OTUB1 promotes metastasis and serves as a marker of poor prognosis in colorectal cancer. Mol Cancer 2014; 13:258; PMID:25431208; http://dx.doi.org/10.1186/1476-4598-13-258
- Karunarathna U, Kongsema M, Zona S, Gong C, Cabrera E, Gomes AR, Man EP, Khongkow P, Tsang JW, Khoo US, et al. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene 2016; 35:1433-44; PMID:26148240; http://dx.doi.org/10.1038/onc.2015.208
- Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 2016; 26:484-98; PMID:27002218; http://dx.doi.org/10.1038/cr.2016.31
- Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol 2016; 6:7; PMID:26858935; http://dx.doi.org/10.3389/fonc.2016.00007
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/10.1038/nature11252
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513:202-9; PMID:25079317; http://dx.doi.org/10.1038/nature13480
- Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497:67-73; PMID:23636398; http://dx.doi.org/10.1038/nature12113
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016; 48:607-16; PMID:27158780; http://dx.doi.org/10.1038/ng.3564
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486:400-4; PMID:22722201