
Mutations in the p53 tumor suppressor gene are the most common genetic alteration in cancer cells.Citation1 In normal cells, wild-type p53 responds to DNA damage and many other types of cellular stress that challenge tissue homeostasis. As a bona fide transcription factor, p53 induces complex gene expression programs that stall progression of the cell cycle until damage is repaired or trigger apoptosis if the stress is too severe to be resolved.Citation2 Different from other tumor suppressors p53 is rarely affected by non-sense or frameshift mutations that provoke a premature termination of translation and would, in principle, be equally effective at disrupting its tumor suppressor activity. Instead, missense mutations are selected during clonal tumor evolution.Citation1 It is therefore believed that missense mutations provide the mutant with de novo oncogenic functions that drive cancer development and progression. As tumors frequently become addicted to the presence of such gain-of-function (GOF) p53 mutants the underlying mechanisms are of intense therapeutic interest.
In a recent article, Vogiatzi et al. used comparative gene expression profiling to identify ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5) as a specific target gene of GOF mutant p53.Citation3 The finding is remarkable for at least 2 reasons. First, ENTPD5 expression is not affected by wild-type p53, thereby providing supporting evidence for the GOF concept. Second, ENTPD5 is upregulated by various p53 GOF mutants in a wide range of cancer cell lines. In addition, the expression level of ENTPD5 in human tumors from various tissue origins correlates with the presence of p53 GOF mutants. These observations suggest that the upregulation of ENTPD5 by GOF mutant p53 may be a rather common recurrent theme.
ENTPD5 belongs to the calnexin/calreticulin chaperon system that assists folding of newly synthesized N-glycosylated proteins in the endoplasmic reticulum (ER) ().Citation4,5 The N-glycosylation of proteins initiates cotranslationally in the ER by en bloc transfer of a complex oligosaccharide to an asparagine residue of the nascent protein. Following an initial trimming process by glucosidases, a single remaining glucose residue constitutes a receptor for the lectins calnexin and calreticulin that assist in the correct folding of the newly synthesized protein. Removal of the glucose residue by glucosidase II releases the protein, now en route to the trans Golgi network for maturation of its N-glycosylated moiety, unless it is recognized as still un-properly folded by the enzyme UDP-glucose:glycoprotein glycosyltransferase (UGGT) that adds a new glucose residue to the oligosaccharide, thereby initiating another round of calnexin/calreticulin-dependent folding. UGGT uses UDP-glucose as a substrate and releases UDP after the enzymatic reaction. ENTPD5 hydrolyzes the released UDP to UMP which is exported from the ER through an antiporter in exchange for UDP-glucose. Thus, the extent of ENTPD5 activity determines the efficiency of the calnexin/calreticulin cycle.
Figure 1. Role of mutant p53 in driving N-glycoprotein folding in the endoplasmic reticulum via the calnexin/calreticulin cycle. mutp53, mutant p53; ENTPD5, ectonucleoside triphosphate diphosphohydrolase 5; UGGT, UDP-glucose:glycoprotein glycosyltransferase; GI, glucosidase I; GII, glucosidase II; CNX, calnexin; CRT, calreticulin; UMP, uridine monophosphate; UDP, uridine diphosphate; red triangle, glucose; ERAD, ER-associated degradation; UPR, unfolded protein response; Glc, glucose; Man, mannose; GlcNAc, N-acetylglucosamine.
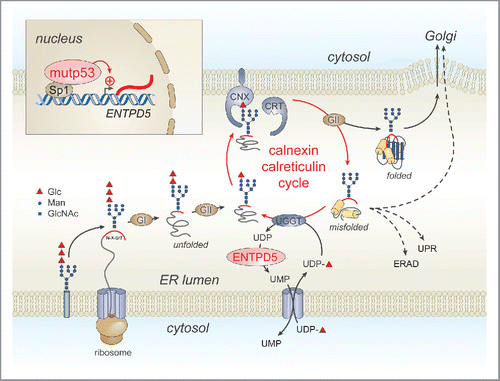
Mutant p53 is known to promote invasiveness and metastasis of cancer cells through upregulation of several receptor tyrosine kinases and integrins at the cell surface, including transforming growth factor β (TGFβ) receptor, platelet-derived growth factor receptor (PDGFRβ), epidermal growth factor receptor (EGFR) and c-MET.Citation1 These receptors are highly N-glycosylated and their maturation likely depends on the calnexin/calreticulin chaperon system.Citation4,6 Thus, overexpression of these surface proteins will stimulate invasion and metastasis only if they are correctly folded. Calnexin/calreticulin cycle deficiency or blockade through genetic deletion or pharmacological inhibition of glucosidase II results in a compromised glycoprotein quality control system, with incompletely folded proteins being either secreted, retained in the ER or prematurely degraded.Citation4 Similarly, ENTPD5 knock-down was shown to decrease the total amount of surface receptors, associated with the induction of an ER stress response and a dramatic attenuation of cell growth.Citation5 Vogiatzi et al. report that downregulation of either mutant p53 or ENTPD5 leads to an accumulation of immature oligosaccharide side chains on endoglin – a pro-metastatic TGFβ co-receptor – reflecting a less active calnexin/calreticulin-dependent quality control.Citation3 Thus, mutant p53, through the regulation of ENTPD5 expression, enhances the efficiency of the N-glycoprotein folding machinery.
Vogiatzi et al. also provide evidence that the regulation of ENTPD5 by mutant p53 has important consequences for cancer cell behavior, since downregulation of ENTPD5 restricts the invasiveness and metastasis potential of cancer cells to a similar extent as the downregulation of mutant p53 itself. Moreover, the reduction of invasion and metastasis observed upon depletion of mutant p53 could be rescued by stable expression of ectopic ENTPD5, indicating that ENTPD5 alone can compensate for the defects observed upon downregulation of mutant p53.
In perspective, the role of ENTPD5 in N-glycoprotein folding may provide new treatment opportunities for the aggressive subtypes of p53 mutant tumors. First, engineered mice lacking the Entpd5 gene are viable and display pathologies only after one year of age, thereby promising a sufficiently broad therapeutic window. Second, ENTPD5 is an enzyme and as such considered druggable. Of note, other components of the glycoprotein folding machinery may also be putative targets and several inhibitors have been developed as antiviral therapies.Citation7 It will therefore be exciting to explore potential anti-metastatic effects of compounds that interfere with N-glycoprotein folding in the ER.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
References
- Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25(3):304-17; PMID:24651012; http://dx.doi.org/10.1016/j.ccr.2014.01.021
- Schlereth K, Charles JP, Bretz AC, Stiewe T. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle 2010; 9(20):4068-76; PMID:20948308; http://dx.doi.org/10.4161/cc.9.20.13595
- Vogiatzi F, Brandt DT, Schneikert J, Fuchs J, Grikscheit K, Wanzel M, Pavlakis E, Charles JP, Timofeev O, Nist A, et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc Natl Acad Sci USA 2016; 113(52):E8433-42; PMID:27956623; http://dx.doi.org/10.1073/pnas.1612711114
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 2004; 73:1019-49; PMID:15189166; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073752
- Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, Wang X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010; 143(5):711-24; PMID:21074248; http://dx.doi.org/10.1016/j.cell.2010.10.010
- Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007; 129(1):123-34; PMID:17418791; http://dx.doi.org/10.1016/j.cell.2007.01.049
- Chang J, Block TM, Guo J-T. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral Res 2013; 99(3):251-60; PMID:23816430; http://dx.doi.org/10.1016/j.antiviral.2013.06.011