
ABSTRACT
Double strand break lesions, the most toxic type of DNA damage, are repaired primarily through 2 distinct pathways: homology-directed recombination (HR) and non-homologous end-joining (NHEJ). BRCA1 and 53BP1, 2 proteins containing the BRCT modular domain, play an important role in DNA damage response (DDR) by orchestrating the decision between HR and NHEJ, but the precise mechanisms regarding both pathways are not entirely understood. Previously, our group identified a putative interaction between BRCA1 and BARD1 (BRCA1-associated RING domain 1) and the cyclin-dependent kinase (CDK9). CDK9 is a component of the positive transcription elongation complex and has been implicated in genome integrity maintenance associated with the replication stress response. Here we show that CDK9 interacts with endogenous BRCA1 and BARD1 mediated by their RING finger and BRCT domains, and describe CDK9 ionizing radiation-induced foci (IRIF) formation and its co-localization with BRCA1 in DNA damage sites. Cells lacking CDK9 are characterized by an altered γ−H2AX foci dynamics after DNA damage, a reduced efficiency in HR but not in NHEJ repair, failure to form BRCA1 and RAD51 IRIF and increased sensitivity to genotoxic agents. These data indicate that CDK9 is a player in the DDR and is consistent with its participation in HR pathway by modulating BRCA1 response.
Introduction
Human cells are constantly exposed to different DNA damaging insults. The DNA damage response (DDR) is a major feature in maintaining genome integrity and consequently suppressing tumorigenesis. DNA double-strand breaks (DSBs) are the most toxic kind of DNA damage and are primarily repaired through 2 major pathways, the error-free homologous recombination (HR) and error-prone non-homologous end joining (NHEJ).Citation1 Both processes rely on the recruitment of different protein complexes that target the DNA repair.Citation2
BRCA1 is a tumor suppressor that plays an essential role in the DDR through both HR and NHEJ by acting as a scaffold protein in different complexes.Citation2-4 The decision between HR and NHEJ is taken based on the recruitment balance of BRCA1 and 53BP1.Citation2,5 53BP1 and its interaction partners RIF1 and PTIP, repress the HR through the inhibition of BRCA1 recruitment to DNA damage sites.Citation5-7 BRCA1 interacts with CtIP, stimulating the 5′end resection and as consequence the HR.Citation7,8 BRCA1-deficient cells present reduced HR repair efficiency that can lead to generate synthetic lethality toward PARP inhibitors, which is actually been used in anticancer therapeutic strategies.Citation9,10 BRCA1 acts commonly associated with BARD1 (BRCA1-associated RING domain protein 1) through dimerization of their RING-finger domains.Citation11 BARD1 protein structure resembles BRCA1, besides their RING-finger domains both proteins enclose tBRCT (tandem BRCT) domains. The tBRCT is present in several proteins involved in the DDR, operating as a protein-protein interaction module capable of recognizing phosphorylated proteins.Citation12,13
In a previous work, our group generated a human protein-protein interaction network centered on interactions mediated by the tBRCT domain and identified CDK9 (cyclin-dependent kinase 9) as a putative interaction partner of BRCA1 and BARD1 tBRCTs.Citation14 The cyclin-dependent kinases (CDKs) participate in many DDR-related processes, such as signal transduction and cell cycle arrest.Citation15,16 CDK9 was originally characterized as the catalytic subunit of the positive elongation complex (P-TEFb), which is responsible for promoting transcription through the phosphorylation of the C-terminal domain (CTD) of the holoenzyme RNA polymerase II.Citation17 Moreover, CDK9 is reported to be involved in genome integrity maintenance, taking part in replication stress response together with ATR and ATRIP.Citation18,19
In this report, we analyze the role of CDK9 in the DDR in association with BRCA1 and BARD1. We demonstrate that the absence of CDK9 lead to a disturbed DDR, showing that CDK9 modulates BRCA1 ionizing radiation-induced foci (IRIF) formation but not 53BP1 and that CDK9-silenced cells present reduced HR (but not NHEJ) repair efficiency, leading to a DNA damage sensitive phenotype in the presence of olaparib, a PARP inhibitor.
Results
CDK9 interacts with BRCA1 and BARD1
Previously, CDK9 was identified as an interaction partner of the tandem BRCT domains of BRCA1 in a yeast 2-hybrid screening. Furthermore, CDK9 was also found to interact with the tandem BRCT domains of BARD1 in a tandem affinity purification assay.Citation14 To confirm these observations, co-immunoprecipitation assays were conducted using nuclear extracts of human cell lines. As shown in , CDK9 was co-immunoprecipitated with endogenous BRCA1 in HeLa cell extracts, and, similarly, BARD1 was identified in the same complex with CDK9 in HEK293FT (), confirming the interaction detected using epitope-tagged tBRCT baits.Citation14 We also conducted reciprocal co-immunoprecipitation of endogenous BRCA1 and BARD1 in HEK293FT cell extracts and confirmed this interaction (Supplementary Figs 1A and 1B).
Figure 1. CDK9 is a new interaction partner of BRCA1 and BARD1. Protein levels were determined in (A) HeLa and (B) HEK293FT nuclear extracts by immunoblotting using specific antibodies. Co-immunoprecipitation assays were performed using anti-CDK9 or anti-HA (IgG) antibodies, immunoblots were developed using anti-CDK9 and anti-BRCA1 or BARD1 antibodies, as indicated. ¥ indicate a non-specific band. (C) Diagram of constructs used to map the BRCA1 interaction with CDK9. RING, RING finger domain; NLS, nuclear localization signals; CC, Coiled-coil domain; tBRCT, tandem BRCT. (D) Upper panels, co-expression of GST-fragments of BRCA1 and FLAG-CDK9 in HEK293FT cells. The lower molecular weight band presented by the empty vector (EV) transfection corresponds to GST. Lower panels, GST pull-down assay, Western blots (WB) were developed using indicated antibodies. (E) Diagram of GST-tagged constructs of BARD1 interaction with CDK9. ANK, ankyrin repeats. (F) Left panels, input of bacterially expressed BARD1 GST-fragments and FLAG-CDK9 produced in HEK293FT cells. The lower molecular weight band presented by the empty vector (EV) transfection corresponds to GST. Right panels, GST pull-down assay, WBs were developed using indicated antibodies. * indicates fragments of interest.
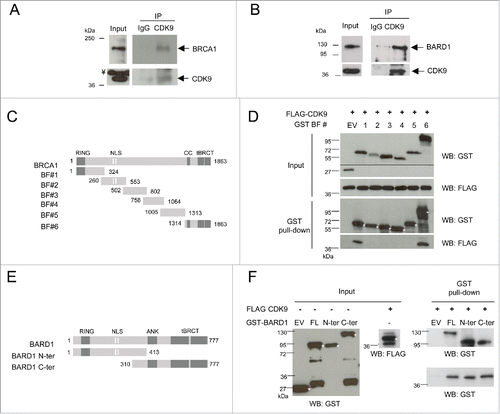
Next, we analyzed whether other regions, besides the tandem BRCTs, were responsible for mediating the interaction of CDK9 with BRCA1 and BARD1. For this purpose, BRCA1 GST-tagged fragments () were expressed in HEK293FT cells and used as baits for pulldown assays against FLAG-tagged CDK9. Both the C-terminal (including the coiled-coil and the tBRCT domains; fragment BF#6) and the amino-terminal region (including the RING-finger domain; fragment BF#1) of BRCA1 were shown to be sufficient for CDK9 interaction ().
We also investigated whether BARD1 N- and C- terminal regions were capable of interacting with CDK9. As presented in , both regions of BARD1 (GST-tagged fragments, ) interact with CDK9. Taken together, these data indicate that CDK9 interacts with both BRCA1 and BARD1 through interactions in different domains.
CDK9 is involved in the DNA damage response
To investigate the involvement of CDK9 in the DDR, MCF7 cells were stably silenced for CDK9 (MCF7 shCDK9) and for a non-target scrambled shRNA control (MCF7 shSCR). As presented in , CDK9 protein level is nearly undetectable in MCF7 shCDK9 nuclear extracts. It is worth of note that CDK9 silencing did not affect BRCA1 and BARD1 expression despite its involvement in transcription regulation ().
Figure 2. CDK9-silenced cells present an altered DNA damage response. (A) CDK9, BRCA1 and BARD1 expression profile in MCF7 shSCR (negative control) and MCF7 shCDK9 nuclear extracts. TBP was used as loading control. ¥ indicate a non-specific band. (B) γH2AX foci formation dynamics, CDK9-silenced cells were exposed to IR (5 Gy) and immunostained after the indicated time intervals using anti-phosphorylated H2AX (Ser139). Scale bar = 10 μm (C) Phosphorylated H2AX (Ser139) foci were quantified using Image J software. Data is presented as mean ± SD of positive nuclei with 5 or more foci. (D) HR and NHEJ repair efficiency quantification in cells lacking CDK9 expression. Cells were analyzed 72 hours after co-transfection of linearized reporter plasmids (HR or NHEJ) and the DsRed expression vector. Data is presented as mean ± SD of percentage of GFP positive cells relative to DsRed positive cells.
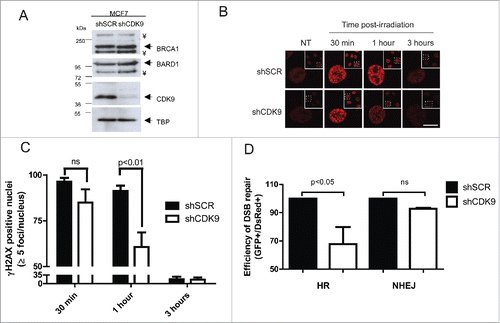
During the DDR, histone H2AX is rapidly phosphorylated (γH2AX) after DSBs and acts as a signal for recruitment of different complexes that are responsible for the DNA damage repair.Citation20,21 Thus, we decided to investigate the dynamics of DNA damage repair through γH2AX foci status in MCF7 shSCR and shCDK9 cells after ionizing radiation (IR) treatment (). Cells lacking CDK9 expression presented a slightly reduction in positive nucleus (84.9% with more than 5 foci) when compared with proficient cells (96.4%) in 30 min after 5 Gy treatment. In 1 hour post-treatment, shCDK9 cells exhibited almost one third less positive nucleus than control cells (60.6% versus 91.3%). After 3 hours, no statistically significant difference was observed (16% vs. 17%). Similar data were obtained using an independent CDK9 shRNA (shCDK9–2), as shown in Supplementary Figures 1C-E.
DNA DSBs are mostly repaired through 2 distinct pathways (HR and NHEJ) and BRCA1 is known for playing a pivotal role in coordinating both.Citation1-4 To investigate the involvement of CDK9 in these pathways we examined the HR and NHEJ repair status using specific I-SceI cleavage dependent reporter assays.Citation22 As shown in , cells lacking CDK9 expression exhibited an impaired DNA damage repair represented by a reduction of 32.2% in HR repair efficiency while NHEJ was not altered. Similar results were observed using MCF7 cells silenced with the shCDK9–2 (Supplementary Figure 1F). These suggest that CDK9 may play a role in the DNA damage repair through HR.
CDK9 IRIF formation and BRCA1 co-localization
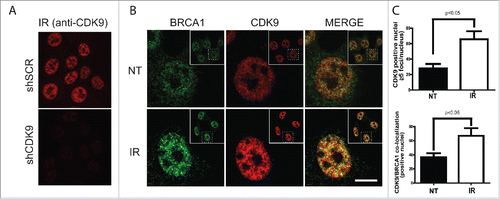
IR-induced foci formation is a classical phenotype of DDR-related proteins and is a consequence of their recruitment and accumulation at DSBs sites.Citation23 CDK9 interaction with BRCA1 and BARD1, key players in the DDR, led us to inquire whether CDK9 was recruited to DNA damage sites and form IRIF. To address this question, we first analyzed the nuclear distribution of CDK9 in IR-treated MCF7 cells. As shown in , non-irradiated MCF7 cells presented a CDK9 diffuse nuclear distribution, while in IR-treated cells CDK9 accumulates in ionizing radiation-induced foci, representing more than 2-fold increase in CDK9 positive nuclei when compared with non-treated cells (). Additionally, we also demonstrated that CDK9 co-localizes with BRCA1 () and RPA at DSB sites (Supplementary Fig 2A). These data suggest that CDK9 is recruited to IR-induced DSB sites and could act in association with BRCA1 in the DNA damage signaling.
Figure 3. CDK9 IRIF co-localize with BRCA1 at damaged DNA sites. MCF7 cells were exposed to IR (10 Gy) and recovered for 3 hours. Immunostaining was performed using (A) only anti-CDK9 or (B) anti-CDK9 and anti-BRCA1 antibodies. Insets depict the nucleus in lower magnification. ((C)- upper panel) Quantification of CDK9 positive nuclei and ((C)- lower panel) CDK9/BRCA1 co-localization.NT, not treated. Scale bars = 10 μm.
CDK9 modulates BRCA1 recruitment to DSBs
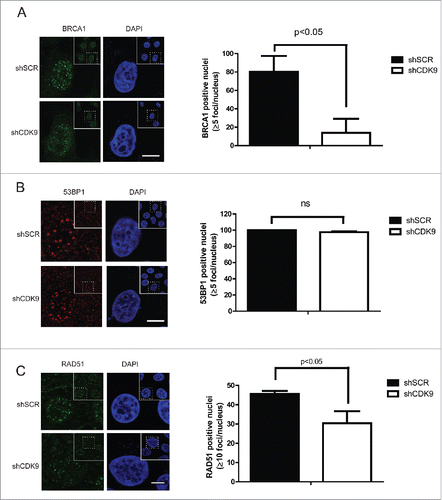
BRCA1 and 53BP1 are determinants of the choice between HR and NHEJ; while BRCA1 acts by favoring HR, 53BP1 stimulates NHEJ.Citation5-8 During the HR pathway BRCA1 is recruited to the DSB sites conducting repair through different complexes.Citation29 Taking into account CDK9 co-localization with BRCA1 at DNA damaged sites, we decided to investigate the role of CDK9 on BRCA1 IRIF formation using confocal microscopy (). In response to DNA damage, MCF7 shSCR cells displayed well-characterized BRCA1 foci formation,Citation24,25 but this phenotype was not observed in the absence of CDK9. MCF7 shCDK9 presented almost 6-fold reduction in positive nucleus (13.8%) than shSCR cells (80.2%). Similar result was observed in shCDK9–2 cells (Supplementary Fig. 2B). Next, we analyzed 53BP1 foci formation in MCF7 shSCR and shCDK9 cells (). CDK9 deficient cells exhibited a similar recruitment profile as control cells. Corroborating these data, the recruitment of the recombinase RAD51 was also reduced in cells lacking CDK9 expression when compared with control cells (one third less in shCDK9 cells when compared with shSCR) ( and Supplementary Fig. 2C). Collectively, these data strongly suggest that CDK9 is required for the localization of BRCA1 and consequently RAD51, but not 53BP1 to DSB sites.
Figure 4. BRCA1 and RAD51, but not 53BP1, recruitment to damaged DNA sites are dependent upon CDK9. MCF7 shSCR or shCDK9 cells were exposed to IR (10 Gy) and recovered for 3 hours (A and B) or for 5 hours (C). Left, immunofluorescence staining using anti-BRCA1, anti-53BP1 or anti-RAD51. Scale bars = 10 μm. Right, BRCA1, 53BP1 and RAD51 foci quantification using Image J software. Data is presented as mean ± SD.
Absence of CDK9 sensitizes cells to DNA damage
Dysfunctional DNA repair is usually followed by an increase in sensitivity to DNA damaging agentsCitation26-28 We performed cellular survival assays to explore whether the impaired recruitment of BRCA1 to DSB sites in the absence of CDK9 could sensitize cells to IR treatment. As shown in , a radiosensitive phenotype was demonstrated in 2 independent MCF7 CDK9 silenced cell lines. Statistically significant differences were observed in 2, 3 and 4 Gy treatment. This observation is supported by evidences described by Storch and Cordes showing radiosensitivity associated to CDK9 depletion in human head and neck squamous cell carcinoma (HNSCC) cell lines.Citation30
Figure 5. Cells lacking CDK9 exhibit increase sensitivity to genotoxic agents. (A) MCF7 shSCR, shCDK9 and shCDK9–2 were subjected to long-term clonogenic assay after irradiation (1, 2, 3 or 4 Gy). Cells were fixed and stained with crystal violet and colonies were quantified. (B) Cells were also subjected to viability assay after treatment with ionizing radiation in the presence or not of PARP inhibitor, olaparib (50 nM). Viability quantification were determined by the absorbance of crystal violet at 590 nm. Data are presented as means ± SD of triplicates. *** = p<0.001 and ** = p<0.01
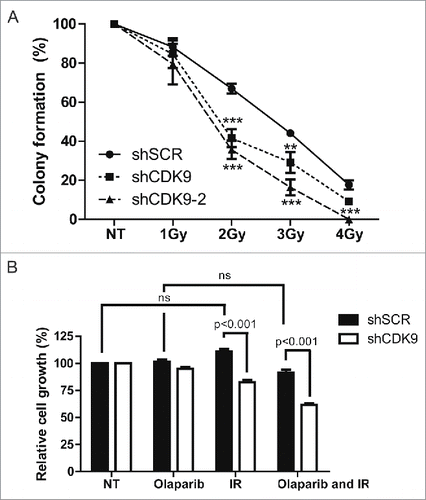
Inhibition of PARPs is constantly associated with synthetic lethality in cells with compromised HR repair.Citation9,10 Thus, we decided to evaluate whether cells lacking CDK9 expression were more sensitive to DSB in the presence of the PARP inhibitor olaparib. As shown in , IR and olaparib combined treatment lead to an additive effect in cells lacking the CDK9 expression, inhibiting cell growth (61.5%) when compared with proficient cells (91.3%). These data corroborate a possible involvement of CDK9 in DSB repair through HR.
Discussion
CDK9 is described as the catalytic subunit of the P-TEFb complex and has been implicated in different biologic processes such as transcription regulation, mRNA splicing and HIV replication.Citation31,32 Yu and colleagues depicted CDK9 as an interaction partner of ATR and ATRIP and in its absence cells were more sensitive to hydroxyurea. The scenario is very suggestive to CDK9 participation in genome integrity maintenance through the replication stress response (RSR).Citation18 Moreover, SIRT2 was described directing the RSR through CDK9 deacetylation, confirming the idea that CDK9 is involved in the RSR.Citation19
Here we demonstrate that CDK9 is an interaction partner of BRCA1 and BARD1 in human cells. BRCA1 and BARD1 are well-characterized players in the DDR.Citation33,34 Our data indicate that BRCA1 and BARD1 interact with CDK9 by their N- and C-terminal regions (RING-finger and the tBRCT; ), which corroborates our previous report that described CDK9 as a putative interaction partner of the tBRCT domains of BRCA1 and BARD1.Citation14 Like this, CDK9 is present in complexes with BRCA1 and BARD1, raising the hypothesis of its participation in the DDR.
Upon DNA damage, ATM rapidly phosphorylates H2AX, which acts as a critical DSB marker for signal transduction and the hierarchal recruitment of proteins that dictate whether HR or NHEJ would repair the DNA damage.Citation2,20,21
Supporting the hypothesis of a CDK9 role in the DDR, we demonstrated that CDK9-silenced cells exhibit an altered γH2AX foci formation dynamics despite the fact that BRCA1 and BARD1 expression were not affected by CDK9 knockdown (). In this context, we also observed that DNA repair efficiency carried by HR, but not NHEJ, was reduced in shCDK9 cells, placing CDK9 as an actor in the HR pathway.
BRCA1 is a central protein during the DNA damage repair, operating in different steps in HR, such as DNA end resection, signal transduction and RAD51 loading on the single strand DNA (ssDNA).Citation7,8,37-39 BRCA1 tBRCT domain is responsible for promoting protein-protein interactions, mediating the recruitment and retention at the DSB sites.Citation35,36 During the DNA repair signaling, as DDR-related proteins are recruited to DSB sites, they accumulate and can be visualized as nuclear foci.
We performed the first observation of CDK9 IRIF formation and its co-localization with BRCA1 and RPA at DNA damaged sites ( and Supplementary Fig 2), endorsing a CDK9 possible role in HR repair.
The molecular basis of the choice between HR and NHEJ could be summarized by the efficient recruitment of BRCA1 or 53BP1 to the DSB site, modulating the extension of DNA end resection.Citation5-8 We inquired whether the participation of CDK9 in HR could be related to the regulation of BRCA1 and 53BP1 recruitment to damaged sites. Interestingly, CDK9-silenced cells presented a drastic reduction of BRCA1 (but not 53BP1) IRIF formation (). As a downstream event, RAD51 loading on chromatin was also observed impaired (), suggesting that BRCA1 recruitment to DSBs is dependent upon CDK9 and consequently for HR repair.
IR treatment may also promote SSBs, which are repaired in a PARP1/2-dependet mechanism.Citation9 In cycling cells, if not repaired, SSBs tend to progress to DSBs that would be preferentially repaired through HR.Citation40,41 Consistent with our findings, CDK9-silenced cells exhibited an increased sensitivity to IR treatment that was intensified in the presence of a PARP inhibitor ().
Ultimately, we propose a model where CDK9 acts in the HR pathway by promoting BRCA1 recruitment to DSB sites. However, further investigation is required to characterize how CDK9 recruitment occurs and whether its absence is sufficient for impairment of other BRCA1 functions.
Materials & methods
Cell culture and antibodies
The human mammary gland carcinoma MCF7 and the cervix adenocarcinoma HeLa cell lines were obtained from the ATCC Cell Bank. Human HEK293FT cell line was purchased from Invitrogen (Invitrogen). Cells were maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (Invitrogen) in 5% v/v CO2 at 37°C.
Rabbit anti-BARD1 (Bethyl Laboratories, BL518), rabbit anti-CDK9 (Bethyl laboratories, A303–493A), rabbit anti-HA (Santa Cruz Biotech, SC-805) and rabbit anti-BRCA1 (Santa Cruz Biotech, SC-642) antibodies were used for co-immunoprecipitation and immunoblotting. Mouse anti-FLAG (Sigma Co., F1804), mouse anti-β-actin (Santa Cruz Biotech, SC47778), mouse anti-GST (Santa Cruz Biotech, SC-138), goat HRP-conjugated anti-mouse Ig (Santa Cruz Biotechnologies, SC2005) and goat HRP-conjugated anti-rabbit Ig (Santa Cruz Biotechnologies, SC2301) antibodies were used for immunoblotting. Rabbit anti phospho-H2AXSer139 monoclonal antibody (Santa Cruz Biotech, SC-101696), mouse anti-BRCA1 (Calbiochem, OP107), mouse anti-RAD51 (Thermo Fisher, 3C10), rabbit anti-CDK9 (Bethyl laboratories, A303–493A), rabbit anti-53BP1 (Bethyl laboratories, A300–272A), rat anti-RPA32/30 (Cell Signaling 2208), goat Alexa Fluor 488 conjugated anti-rabbit Ig antibody (Life Technologies, A11008), rabbit biotinylated anti-rat (Vector Labs, BA-4000), Streptavidin-Cy3 (Thermo Fisher, 434315) and goat Alexa Fluor 546 conjugated anti-rabbit Ig antibody (Life Technologies, A-11035) were used for immunofluorescence staining. All antibodies were used following manufacturer's instructions.
Constructions and transfection
Human CDK9 coding sequence (PubMed Accession Number: NM_001261.3) was obtained by PCR amplification using the following primers CDK9-Fw (5′-AAGAATTCAATGGCAAAGCAGTACGACTCGGTG-3′, enclosing EcoRI restriction site) and CDK9-Rv (5′-AAGGATCCTCAGAAGACGCGCTCAAACTCC-3′, enclosing BamHI restriction site) and the construction pCDNA3-CDK9-HA as template (purchased from Addgene; Plasmid #635). The amplified coding sequence was cloned into pCMV2-FLAG (Sigma). pGEX6p1-BARD1 constructs, were generated by the same approach described above, using the construction pYFP-BARD1 (kindly gift from Dr Beric Henderson) as template and the following primers: BARD1 N-ter Fw (5′-AAGTCGACAATGCCGGATAATCGGCAGC-3′) and Rv (5′-AAGCGGCCGCTCAATGGAGCAAAGTCTCTCCT-3′), BARD1 C-ter Fw (5′- AAGTCGACAAAGTCCCATTTCTAAGAGATGTAGAAC-3′ and Rv (5′-AAGCGGCCGCTCAGCTGTCAAGAGGAAGCAAC-3′). pEBG-BRCA1 constructs were a kindly gift from Dr Toru Ouchi.
Transfections were conducted using Polyethylenimine (PEI; Polysciences Inc, Pennsylvania, EUA) as described previously.Citation42
GST pulldown, co-immunoprecipitation and immunoblotting
GST-BARD1 fragments were obtained from bacterial extracts following GE Gene Fusion System Handbook.Citation43 Briefly, E. coli (BL21 strain) cells previously transformed with BARD1 pGEX constructs were treated with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 hours at 30°C. Then, cells were submitted to sonication and cellular debris were removed by centrifugation, the supernatant enriched with GST-proteins was recovered. Mammalian total cellular extracts and nuclear extracts were obtained as described by Carvalho et al.Citation44
GST pulldown assays were performed by incubating Glutathione Sepharose 4B (GE Healthcare) with proper cellular extracts (derived from bacterial and/or mammalian cells) for 16 h at 4°C followed by extensively buffer washes. All incubations and washes were performed using ice-cold mild-RIPA buffer supplemented with 2.5 mM dithiothreitol (DTT).
Co-immunoprecipitation assays were performed by incubating A/G plus agarose beads (Santa Cruz Biotech), cellular extracts and the appropriate antibody for 16 h at 4°C in mild-RIPA buffer, followed by extensively ice-cold mild-RIPA buffer washes. Immunoblottings were performed using PVDF membranes (Millipore) and developed using ECL Plus kit (Amersham Biosciences).
CDK9 silencing
Lentiviral particles enclosing pLKO.1 plasmids encoding shRNAs targeting CDK9 gene (shCDK9 – Openbiosystem plasmids TCRN#494 and TCRN#497) or a control scrambled sequence (shSCR – Openbiosystem plasmid RHS6848) were produced in HEK293FT cells using ViraPower Lentiviral Expression Kit (Invitrogen). To generate MCF7 shCDK9 and shSCR cells, lentiviral particles were transduced in MCF7 cells, followed by puromycin (Invitrogen) selection according to manufacturer instructions.
Immunofluorescence
Immunofluorescence staining was performed as described previously by Sy et al.,Citation45 with slight modifications. Briefly, cells were plated over glass covers slides and allowed to attach for 24 h. Afterwards, cells were exposed to IR (5 or 10 Gy) and at the indicated time point were submitted to fixation (20 minutes in 4% w/v formaldehyde prepared in PBS). Cells were incubated with anti-BRCA1, anti-RPA or anti-CDK9 followed by anti-mouse conjugated with Alexa Fluor 488 or anti- rabbit conjugated with Alexa Flour 546 or biotinylated anti-RAT followed by Streptavidin-Cy3, respectively. For 53BP1, RAD51 and phospho-H2AXser139 staining cells were pre-treated with cytoskeleton buffer (10 mM HEPES/NaOH pH 7.4, 300 mM sucrose, 100 mM NaCl and 3 mM MgCl2) supplemented with 0.5% v/v triton X-100 for 2 min on ice.52 Slides were mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). Samples were analyzed by confocal microscopy (Olympus FV10i). IRIF were quantified using ImageJ software (NIH).
Analysis of DNA damage repair in mammalian cells
HR and NHEJ DNA repair efficiency were assessed as described previously by Mao et al.Citation22 Briefly, MCF7 shSCR and MCF7 shCDK9 cells were plated and allowed to attach for 24 h. Afterwards, cells were transfected with I-SceI digested HR or NHEJ reporter plasmids. Three days later, cells were harvested and washed with PBS solution. Samples were acquired using flow cytometry (Accury C6 BD Biosciences) and analyzed with CFlow® plus v1.0.227.4 (Accury® Cytometers, Inc.).
Cellular survival assays
Cells were seeded in 6-well plates (1×102 cells/well for colony formation or 1×103 cells/well for viability analysis), allowed to attach for 24 h, and then irradiated (1, 2, 3 or 4) treated or not with 1μM olaparib followed by a 7-day recovery period. Cellular viability was assessed using 1% w/v crystal violet staining for 15 min, followed by 10% v/v acetic acid elution and quantification in spectrophotometer at 590 nm wavelength. Colony formation was directly quantified under the stereo microscope (Zeiss).
Abbreviations
| CDK | = | cyclin-dependent kinase |
| CTD | = | C-terminal domain |
| C-ter | = | C-terminal region |
| DDR | = | DNA damage response |
| DSB | = | DNA double-strand break |
| DTT | = | dithiothreitol |
| HR | = | homology-directed recombination |
| IPTG | = | isopropyl β-D-1-thiogalactopyranoside |
| IR | = | ionizing radiation |
| IRIF | = | ionizing radiation-induced foci |
| NHEJ | = | non-homologous end-joining |
| N-ter | = | N-terminal region |
| RSR | = | replication stress response |
| SSB | = | single stranded break |
| ssDNA | = | single stranded DNA |
| tBRCT | = | tandem BRCT domains |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1295177_Supplemental_Material.zip
Download Zip (13.5 MB)Acknowledgments
The authors would like to acknowledge Fundação de Amparo à Pesquisa do Rio de Janeiro – FAPERJ/Brazil, Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPQ/Brazil and the INCA Confocal Microscopy Facility.
Related Research Data
References
- Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47(4):497-510; PMID:22920291; http://dx.doi.org/10.1016/j.molcel.2012.07.029
- Thompson LH. Recognition. signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat Res 2012; 751(2):158-246; PMID:22743550; http://dx.doi.org/10.1016/j.mrrev.2012.06.002
- Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 2014; 34(8):1380-8; PMID:24469398; http://dx.doi.org/10.1128/MCB.01639-13
- Bau DT, Mau YC, Shen CY. The role of BRCA1 in non-homologous end-joining. Cancer Lett 2006; 240(1):1-8; PMID:16171943; http://dx.doi.org/10.1016/j.canlet.2005.08.003
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15(1):7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Escribano-Diaz C, Durocher D. DNA repair pathway choice–a PTIP of the hat to 53BP1. EMBO Rep 2013; 14(8):665-6; PMID:23846307; http://dx.doi.org/10.1038/embor.2013.99
- Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 2013; 288(16):11135-43; PMID:23486525; http://dx.doi.org/10.1074/jbc.M113.457440
- Cruz-García A, López-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep 2014; 9(2):451-9; PMID:25310973; http://dx.doi.org/10.1016/j.celrep.2014.08.076
- Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011; 5(4):387-93; PMID:21821475; http://dx.doi.org/10.1016/j.molonc.2011.07.001
- Aly A, Ganesan S. BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol 2011; 3(1):66-74; PMID:21278454; http://dx.doi.org/10.1093/jmcb/mjq055
- Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev 2002; 12(1):86-91; PMID:11790560; http://dx.doi.org/10.1016/S0959-437X(01)00269-6
- Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D. Structure-function of the tumor suppressor BRCA1. Comput Struct Biotechnol J 2012; 1(1):pii: e201204005; PMID: 22737296
- Mesquita RD, Woods NT, Seabra-Junior ES, Monteiro AN. Tandem BRCT domains: DNA's Praetorian Guard. Genes Cancer 2010; 1(11):1140-6; PMID:21533002; http://dx.doi.org/10.1177/1947601910392988
- Woods NT, Mesquita RD, Sweet M, Carvalho MA, Li X, Liu Y, Nguyen H, Thomas CE, Iversen ES Jr, Marsillac S, et al. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci Signal 2012; 5(242):rs6; PMID:22990118; http://dx.doi.org/10.1126/scisignal.2002255
- Cerqueira A, Santamaría D, Martínez-Pastor B, Cuadrado M, Fernández-Capetillo O, Barbacid M. Overall Cdk activity modulates the DNA damage response in mammalian cells. J Cell Biol 2009; 187(6):773-80; PMID:19995934; http://dx.doi.org/10.1083/jcb.200903033
- Wohlbold L, Fisher RP. Behind the wheel and under the hood: functions of cyclin-dependent kinases in response to DNA damage. DNA Repair (Amst) 2009; 8(9):1018-24; PMID:19464967; http://dx.doi.org/10.1016/j.dnarep.2009.04.009
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 2000; (8):2629-34; PMID:10733565; http://dx.doi.org/10.1128/MCB.20.8.2629-2634.2000
- Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep 2010; 11(11):876-82; PMID:20930849; http://dx.doi.org/10.1038/embor.2010.153
- Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, Duong DM, Park SJ, Kim HS, Vassilopoulos A, et al. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc Natl Acad Sci U S A 2013; 110(33):13546-51; PMID:23898190; http://dx.doi.org/10.1073/pnas.1301463110
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276(45):42462-7; PMID:11571274; http://dx.doi.org/10.1074/jbc.C100466200
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273(10):5858-68; PMID:9488723; http://dx.doi.org/10.1074/jbc.273.10.5858
- Mao Z, Jiang Y, Liu X, Seluanov A, Gorbunova V. DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia 2009; 11(7):683-91; PMID:19568413; http://dx.doi.org/10.1593/neo.09312
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40(2):179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 2012; 125(Pt 15):3529-34; PMID:22553214; http://dx.doi.org/10.1242/jcs.105353
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev 2011; 25(7):685-700; PMID:21406551; http://dx.doi.org/10.1101/gad.2011011
- Jeggo P, Lavin MF. Cellular radiosensitivity: how much better do we understand it? Int J Radiat Biol 2009; 85(12):1061-81
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A 2002; 99(12):8173-8; PMID:12034884; http://dx.doi.org/10.1073/pnas.122228699
- Lim YC, Roberts TL, Day BW, Stringer BW, Kozlov S, Fazry S, Bruce ZC, Ensbey KS, Walker DG, et al. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol Oncol 2014; 8(8):1603-15; PMID:25017126; http://dx.doi.org/10.1016/j.molonc.2014.06.012
- Savage KI, Harkin DP. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J 2015; 282(4):630-46; PMID:25400280; http://dx.doi.org/10.1111/febs.13150
- Storch K, Cordes N. The impact of CDK9 on radiosensitivity, DNA damage repair and cell cycling of HNSCC cancer cells. Int J of Oncol 2016; 48:191-8
- Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, Murphy S. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol 2015; 22(5):396-403; PMID:25849141
- Mancebo HS1, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev 1997; 11(20):2633-44; PMID:9334326; http://dx.doi.org/10.1101/gad.11.20.2633
- Sato K, Sundaramoorthy E, Rajendra E, Hattori H, Jeyasekharan AD, Ayoub N, Schiess R, Aebersold R, Nishikawa H, Sedukhina AS, et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr Biol 2012; 22(18):1659-66; PMID:22863316; http://dx.doi.org/10.1016/j.cub.2012.07.034
- Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, Fletcher A, Blair-Reid S, Beesley J, Johal B, et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 2016; 23(7):647-55; PMID:27239795
- Wu W, Nishikawa H, Fukuda T, Vittal V, Asano M, Miyoshi Y, Klevit RE, Ohta T. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res 2015; 75(7):1311-21; PMID:25634209; http://dx.doi.org/10.1158/0008-5472.CAN-14-2796
- Wu Q, Paul A, Su D, Mehmood S, Foo TK, Ochi T, Bunting EL, Xia B, Robinson CV, Wang B, Blundell TL. Structure of BRCA1-BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol Cell 2016; 61(3):434-48; PMID:26778126; http://dx.doi.org/10.1016/j.molcel.2015.12.017
- Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 2009; 106(17):7155-60; PMID:19369211; http://dx.doi.org/10.1073/pnas.0811159106
- Cousineau I, Abaji C, Belmaaza A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res 2005; 65(24):11384-91; PMID:16357146; http://dx.doi.org/10.1158/0008-5472.CAN-05-2156
- Deng CX. BRCA1 cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 2006; 34(5):1416-26; PMID:16522651; http://dx.doi.org/10.1093/nar/gkl010
- Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol 2015; 12(1):27-41; PMID:25286972; http://dx.doi.org/10.1038/nrclinonc.2014.163
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434(7035):917-21; PMID:15829967; http://dx.doi.org/10.1038/nature03445
- Longo PA, Kavran JM, Kim MS, Leahy DJ. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol 2013; 529:227-40; PMID:24011049
- GE Healthcare Life Sciences. GST Gene Fusion System Handbook. 2002. Available at: http://docplayer.net/147913-Ge-healthcare-life-sciences-gst-gene-fusion-system-handbook-imagination-at-work.html
- Carvalho RS, Fernandes VC, Nepomuceno TC, Rodrigues DC, Woods NT, Suarez-Kurtz G, Chammas R, Monteiro AN, Carvalho MA. Characterization of LGALS3 (galectin-3) as a player in DNA damage response. Cancer Biol Ther 2014; 15(7):840-50; PMID:24755837; http://dx.doi.org/10.4161/cbt.28873
- Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle 2010; 9(7):1421-33; PMID:20305393; http://dx.doi.org/10.4161/cc.9.7.11256