
Haematopoietic stem and progenitor cells (HSPCs), which sustain lifelong production of all haematopoietic cell types, are known to be exquisitely sensitive to DNA-damaging events, like γ-irradiation. Moreover, accumulation of DNA damage has been proposed to critically contribute to age-related decline of HSPC function. In line with this view, mouse mutants carrying genetic defects in any of several different genes involved in the DNA damage response (DDR) exhibit strikingly similar deficits in HSPC function. However, despite comprehensive analysis of several such mouse mutants, our knowledge about the mechanistic links between DNA damage and impaired HSPC function has remained rather rudimentary.
In a recent effort to characterize phenotypic abnormalities in a targeted mouse mutant lacking Mixed-Lineage-Leukemia-5 (MLL5), an understudied SET-domain protein, we noticed striking functional deficits in the HSPC compartment closely resembling those in mouse mutants with a defective DDR.Citation1 Indeed, appropriate assays revealed accrued DNA damage in HSPCs lacking MLL5. Moreover, MLL5-deficient HSPCs had drastically increased levels of ROS, and in vivo anti-oxidant treatment efficiently restored HSPC function, as reported previously for mice lacking Ataxia Telangiectasia Mutated (ATM) kinase, a key player in DDR.Citation2 Taken together, our findings provided yet another example for the toxic liaison between DNA damage, elevated ROS and defective HSPC function (see ), as described initially in Atm−/− mice, and meanwhile confirmed for several other mouse mutants with accrued DNA damage in HSPCs.
Figure 1. The IFN-1 > BID > ROS pathway connects DNA damage with hyper-proliferation and malfunction of haematopoietic stem/progenitor cells (HSPCs). Loss of ATM, loss of ATM-mediated BID phosphorylation and/or DNA damage all result in mitochondrial accumulation of full-length BID and toxic ROS production. How nuclear DNA damage may trigger an IFN-1 response is currently unknown, but innate immune signaling via the cytoplasmic DNA recognition machinery involving cyclic GMP-AMP-Synthase (cGAS), stimulator of IFN-1 signaling (STING) and interferon response factor 3 (IRF3) seems a good bet. The mechanistic link between IFN-1 signaling and BID mobilization has not yet been explored and thus remains entirely obscure.
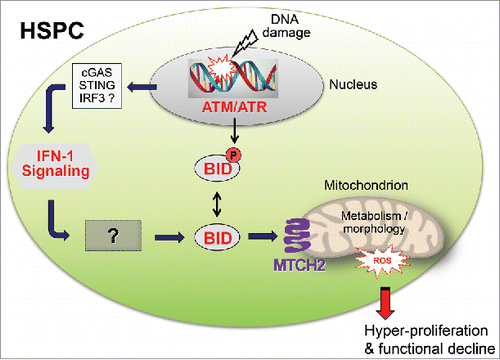
However, analysis of MLL5-deficient HSPCs had more to offer. Several observations, including upregulation of Sca-1, phosphorylation of STAT-1 and transcriptional induction of select IFN-1 target genes, indicated active type I interferon (IFN-1) signaling in HSPCs lacking MLL5.Citation1 Remarkably, genetic abrogation of IFN-1 signaling in Mll5−/− x Ifnar1−/− mice lacking an essential subunit of the IFN-1 receptor reduced ROS to near wild-type levels and markedly restored HSPC function despite absence of MLL5. This striking result identifies IFN-1 signaling as hitherto unknown link between DNA damage and toxic ROS accumulation. Coincidentally, these new findings also provide a lucid explanation for the stunning detrimental effects of IFN-1 induction on HSPC function reported previously.Citation3
But what links IFN-1 signaling with toxic ROS accumulation? Knock-in mice expressing a mutated version of the BH3-only BCL-2-family member BID pointed the way.Citation4 BID was initially shown to connect the extrinsic and intrinsic pathways of apoptosis by being cleaved/activated by Caspase-8 to generate truncated tBID, which then targets mitochondria to induce mitochondrial outer membrane permeabilization (MOMP). MOMP and release of cytochrome c also leads to production of ROS, however this effect was only associated with tBID and induction of apoptosis. Later it was found that full-length BID is phosphorylated by ATM in response to low, non-apoptotic levels of DNA damage, and that this phosphorylation was important for cell cycle arrest at the S phase and inhibition of apoptosis.Citation5 These findings were later challenged by another group, instigating a passionate debate about BID's contribution to the DDR.
To further study these phenomena in vivo, a non-phosphorylatable BID knock-in mouse was generated (BIDS61A/S78A or BIDAA). Strikingly, HSPCs from BIDAA mice also accumulated ROS and exhibited functional deficits virtually identical to those in Mll5−/− and Atm−/− mice.Citation4 Provocatively, loss of BID phosphorylation was associated with marked accumulation of BID at mitochondria in HSPCs from BIDAA and Atm−/− mice, but also in HSPCs from wild-type mice exposed to whole-body irradiation. With these findings in mind, we generated Mll5−/− x Bid−/− compound mice. Importantly, genetic inactivation of Bid resulted in marked reduction of supra-natural ROS levels in Mll5−/− HSPCs and virtually complete rescue of HSPC function. Furthermore, in HSPCs from Mll5−/− mice lacking Ifnar1 mitochondrial BID accumulation was significantly diminished. Taken together, these findings provide conclusive genetic evidence that BID is an important regulator of mitochondrial ROS in the DDR pathway downstream of IFN-1 signaling, and resolves the old controversy about BID's contribution to the DDR.
As usual, new findings raise exciting new questions. For instance, how does BID regulate ROS accumulation in the DDR? A possible candidate for this job was mitochondrial carrier homolog 2 (MTCH2), BID's mitochondrial receptor. MTCH2 was initially shown to play a role in apoptosis: its knockout in the liver significantly reduces tBID-targeting to mitochondria and cytochrome c release, and MTCH2 liver-knockout mice are significantly less sensitive to Fas-induced liver apoptosis in vivo. Interestingly, knockout of MTCH2 in the haematopoietic system triggers a marked increase in mitochondrial metabolism, including an increase in ROS production.Citation6 Thus, MTCH2 appears to act as a regulator of mitochondrial metabolism, and BID is likely to regulate ROS levels via its interaction with MTCH2. Another issue of significant interest pertains to the molecular source of IFN-1 in cells with damaged DNA. Does DNA damage in Mll5−/− mice - and by inference in other mouse mutants with defective DDR - engage the cytosolic DNA sensing machinery of innate immunity, as recent studies in Atm−/− mice suggest?Citation7 Besides, how IFN-1 signaling provokes mitochondrial BID accumulation is an entire mystery. Clearly, the IFN-1 > BID > ROS pathway is just beginning to reveal its secrets.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
H.J.F. received funding through SFB1074-A2 and institutional resources. A.G. is the incumbent of the Marketa and Frederick Alexander Professorial Chair. A.T. was supported by a stipend from the Else-Kröner-Forschungskolleg (EKF).
References
- Tasdogan A, Kumar S, Allies G, Bausinger J, Beckel F, Hofemeister H, Mulaw M, Madan V, Scharfetter-Kochanek K, Feuring-Buske M, et al. DNA damage-induced HSPC malfunction depends on ROS accumulation downstream of IFN-1 signaling and Bid mobilization. Cell Stem Cell 2016; 19:752-67; PMID:27641306; http://dx.doi.org/10.1016/j.stem.2016.08.007
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004; 431:997-1002; PMID:15496926; http://dx.doi.org/10.1038/nature02989
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009; 458:904-8; PMID:19212321; http://dx.doi.org/10.1038/nature07815
- Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, Lapidot T, Jung S, Gross A. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol 2012; 14:535-41; PMID:22446738; http://dx.doi.org/10.1038/ncb2468
- Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC, Gross A. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 2005; 122:593-603; PMID:16122426; http://dx.doi.org/10.1016/j.cell.2005.06.014
- Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, Porat Z, Golan K, Lapidot T, Gross A. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat Commun 2015; 6:7901; PMID:26219591; http://dx.doi.org/10.1038/ncomms8901
- Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015; 42:332-43; PMID:25692705; http://dx.doi.org/10.1016/j.immuni.2015.01.012