
The vascular endothelium, the inner lining of blood vessels, spans approximately 90,000 km through the human body with a surface area larger than a football field. The endothelium provides not only an anti-thrombotic, anti-coagulatory surface to enable nutrient transport to almost every other cell in the human body, but it also provides paracrine (angiocrine) factors that orchestrate cell behavior in an organ-specific manner. To achieve this, endothelial cells secrete soluble factors or provide surface molecules that instruct the differentiation or proliferation rate of adjacent parenchymal cells, e.g. in niches of the bone marrow.Citation1 The tumor vasculature differs morphologically and functionally from that of the healthy organ. There is increasing evidence that endothelial-derived signals, e.g., the expression of Notch ligands, increase the aggressiveness of tumor cells.Citation2,3
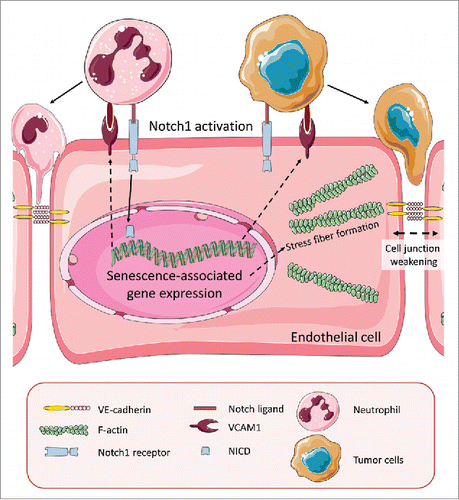
In our recent publication,Citation4 we analyzed a large collection of human cancer samples for the activation rate of Notch1 in the endothelium of tumor blood vessels. Compared to vessels in non-tumorous control tissue, there was a marked increase in Notch1 receptor activation. This correlated with increased metastasis and poor prognosis in humans and mice. Therefore, we asked what consequences a sustained activation of Notch1 would have on endothelial cell behavior. One striking observation was that the overexpression of the constitutively-active Notch1 intracellular domain (NICD) in primary endothelial cells did not only inhibit angiogenesis, but also led to many changes indicative of cellular senescence (proliferation block, fried egg morphology, higher p53 expression, strong expression of senescence-associated β-galactosidase). In addition, endothelial NICD expression increased the synthesis of several chemokines that belong to the senescence-associated secretory phenotype (SASP) and the expression of the adhesion molecule VCAM1. Another feature of endothelial cell senescence is actin stress fiber formation, which we observed upon NICD expression and which led to an increase in the pulling forces toward the cell center. We believe that this pulling force, together with a decrease of VE-cadherin localization at the membrane, led to weakening of cell-to-cell junctions, and thereby to a higher rate of tumor cell transmigration (). In other words, endothelial cell senescence facilitated the entry of tumor cells into the circulation.
Figure 1. Activation of Notch1 receptors in endothelial cells induces senescence. Notch1 receptors on endothelial cells can be activated by Notch ligands expressed by tumor cells or tumor-associated neutrophils. Ligand binding triggers Notch1 cleavage and translocation of the Notch1 intracellular domain (NICD) into the nucleus where it induces expression of senescence-associated genes. Notch1-induced VCAM1 expression promotes adhesion of tumor cells to the endothelium. Endothelial senescence also provokes weakening of cell junctions with diminished VE-cadherin expression and additional mechanical forces by actin stress fibers that pull the membrane toward the cell center. This facilitates transmigration of tumor and immune cells across the blood vessel wall, a crucial step during tumor metastasis.
It is interesting to note that activation of Notch1 receptors and expression of senescence-associated proteins occurred not only within endothelial cells of the primary tumor but also in endothelial cells at distant sites, in particular the lungs. We do not yet know the mechanism as to how this occurs, although it appears likely that tumor or immune cell-secreted Notch ligands in soluble form or incorporated in exsosomes that can travel through the blood stream can activate Notch receptors at distant sites appear most likely. We observed that Notch activation in lung endothelial cells promotes the homing of circulating tumor cells and the infiltration of tumor-associated neutrophils. Both processes were dependent on Notch1-induced expression of the adhesion molecule VCAM1. This indicates that tumors can condition pre-metastatic niches to facilitate their colonization.
Increased endothelial Notch1 activation occurs at sites of atherosclerosis and triggers endothelial senescence and this can promote immune cell infiltration into the atherosclerotic plaque.Citation5 Other reports suggest that hyperglycemia stimulates Notch activity and thereby enhances insulin resistance.Citation6 As such, there is a constantly growing list of pathological conditions that are associated with increased Notch signaling activity and it remains to be seen if some of them can be successfully treated with Notch-inhibiting substances.Citation7
In conclusion, endothelial Notch signaling is not only a central regulator of angiogenesis, but also plays important roles in the resting endothelium. Notch1 over-activation is known to occur in several pathologies. In tumors, sustained endothelial Notch1 signaling induces senescence and changes endothelial behavior in a manner that facilitates several steps of the metastatic cascade.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature 2016; 529:316-25; PMID:26791722; http://dx.doi.org/10.1038/nature17040
- Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 2010; 10:138-46; PMID:20094048; http://dx.doi.org/10.1038/nrc2791
- Cao Z, Ding B Sen, Guo P, Lee SB, Butler JM, Casey SC, Simons M, Tam W, Felsher DW, Shido K, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014; 25:350-65; PMID:24651014; http://dx.doi.org/10.1016/j.ccr.2014.02.005
- Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, Herpel E, Menuchin A, Chang-Claude J, et al. Endothelial Notch1 activity facilitates metastasis. Cancer Cell 2017; 31:355-67; PMID:28238683; http://dx.doi.org/10.1016/j.ccell.2017.01.007
- Liu Z-J, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, Vazquez-Padron RI, Pericak-Vance M, Vance JM, Goldschmidt-Clermont PJ, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012; 2:296-303; http://dx.doi.org/10.1016/j.atherosclerosis.2012.04.010
- Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab 2015; 26:248-55; PMID:25805408; http://dx.doi.org/10.1016/j.tem.2015.02.006
- Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov 2014; 13:357-78; PMID:24781550; http://dx.doi.org/10.1038/nrd4252