
ABSTRACT
Diabetes results from an inadequate mass of functional β cells, due to either β cell loss caused by autoimmune destruction (type I diabetes) or β cell failure in response to insulin resistance (type II diabetes). Elucidating the mechanisms that regulate β cell mass may be key to developing new techniques that foster β cell regeneration as a cellular therapy to treat diabetes. While previous studies concluded that cyclin D2 is required for postnatal β cell self-renewal in mice, it is not clear if cyclin D2 is sufficient to drive β cell self-renewal. Using transgenic mice that overexpress cyclin D2 specifically in β cells, we show that cyclin D2 overexpression increases β cell self-renewal post-weaning and results in increased β cell mass. β cells that overexpress cyclin D2 are responsive to glucose stimulation, suggesting they are functionally mature. β cells that overexpress cyclin D2 demonstrate an enhanced regenerative capacity after injury induced by streptozotocin toxicity. To understand if cyclin D2 overexpression is sufficient to drive β cell self-renewal, we generated a novel mouse model where cyclin D2 is only expressed in β cells of cyclin D2−/− mice. Transgenic overexpression of cyclin D2 in cyclin D2−/− β cells was sufficient to restore β cell mass, maintain normoglycaemia, and improve regenerative capacity when compared with cyclin D2−/− littermates. Taken together, our results indicate that cyclin D2 is sufficient to regulate β cell self-renewal and that manipulation of its expression could be used to enhance β cell regeneration.
Introduction
Diabetes results from an inadequate mass of functional β cells, due to either β cell loss caused by autoimmune destruction (type I diabetes) or β cell failure in response to insulin resistance (type II diabetes).Citation1-3 It may be possible to supplement β cell mass as a cellular therapy by stimulating the self-renewal of pre-existing β cells, by differentiating β cells from multipotent progenitor cells, or by stimulating transdifferentiation from other cell types.Citation4 Self-renewal of pre-existing β cells is the predominant mechanism to expand endogenous postnatal β cell mass in rodents.Citation5 In humans, β cell mass expansion is primarily driven by β cell self-renewal during childhood.Citation6 Although β cell self-renewal drops to very low levels in adulthood, it may be possible to exploit the pathways that regulate β cell self-renewal to drive therapeutic β cell expansion. Thus, understanding the processes that regulates β cell self-renewal may provide novel insights into approaches to expand β cell mass and move the field toward development of novel regenerative therapies for diabetic patients.
β cell self-renewal is dependent on and governed by the precise control of the cell cycle. Entry into the G1 phase of the cell cycle is initiated by D-type cyclins binding to and activating cyclin-dependent kinases. Cyclin D2 is the major D-type cyclin expressed in β cells, and multiple studies have shown its critical requirement for postnatal β cell mass expansion.Citation7-10 However, because these studies did not knockout cyclin D2 specifically in β cells, there was a possibility that unidentified cell types that may compensate for β cell insufficiency by contributing to new β cell formation could also be restricted by the absence of cyclin D2. Evidence for the existence of multipotent progenitor cells in the adult pancreas after severe injury supports the possibility that cyclin D2 maybe required in other compartments of the pancreas that contribute to new β cell formation.Citation11 In addition, overexpression of a stable species of cyclin D2 (T280A) in adult animals increased β cell survival but did not enhance self-renewal, suggesting that extending the half-life of cyclin D2 is not sufficient to enhance β cell mass through self-renewal.Citation12 While the cyclin D2 T280A model illuminated a novel role for cyclin D2 in β cell survival, the analogous phosphorylated form of cyclin D2 has never been detected in β cells, such that the T280A model may not be reflective of how wildtype cyclin D2 may affect β cell self-renewal. Overexpression of wildtype cyclin D2 may have different effects on β cell self-renewal and survival.
To test if the overexpression of wildtype cyclin D2 could stimulate β cell self-renewal, we generated a “knock-in” transgenic mouse that specifically overexpressed cyclin D2 in β cells. We measured a 2-fold increase in cyclin D2 expression in the knock-in β cells, which resulted in an increased β cell mass. β cell-specific overexpression of cyclin D2 extended the ability of postnatal β cells to self-renew post-weaning and enhanced their regenerative capacity in response to injury. To discern if cyclin D2-mediated β cell self-renewal was sufficient to maintain normoglycemia, we bred the β cell-specific cyclin D2 knock-in mice with the global cyclin D2−/− mice. Re-expression of cyclin D2 in cyclin D2−/− β cells restored deficits in cyclin D2−/− β cells mass, re-established the capacity of cyclin D2−/− β cells to respond to glucose challenge, and restored the regenerative capacity relative to cyclin D2−/− littermate mice. These results establish that cyclin D2 is sufficient to drive postnatal β cell self-renewal and can enhance the regenerative capacity of β cells.
Results
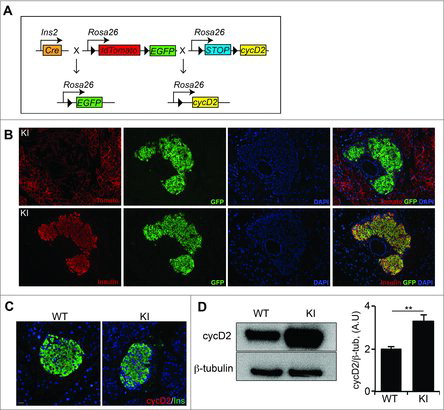
Targeted overexpression of cyclin D2 results in a 2-fold increase in cyclin D2 protein in β cells
Although mice expressing a stable form of cyclin D2 (T280A) revealed a novel role for cyclin D2 in β cell survival, it is not known if the overexpression of native cyclin D2 can specifically drive β cell self-renewal. We generated a transgenic mouse model where cre-recombinase expressed in insulin cells (RIP-cre) drove the overexpression of cyclin D2 and labeled all β cells with a GFP fluorescent lineage trace marker (referred to herein as KI, ). Immunohistochemistry for the GFP protein confirmed efficient cre-mediated recombination by co-expression of GFP and loss of dTomato in insulin-expressing β cells (). Next, we measured the expression of cyclin D2 protein in the WT and KI mice. We and others have reported that the expression of cyclin D2 declines in adult β cells, with a limited number of cells expressing low levels of cyclin D2.Citation7,8 Immunohistochemistry confirmed limited expression in wildtype mice, but revealed brighter cyclin D2 expression in an increased number of β cells in the KI mice (). We used western blot analysis to quantify the abundance of cyclin D2 in islets isolated from 6-week-old mice. Densitometric analysis determined a 2-fold increase in cyclin D2 expression in KI islets compared with islets from WT littermates (). These results suggested that the cyclin D2 knock-in transgene was able to specifically drive the overexpression of cyclin D2 in β cells.
Figure 1. β cell specific overexpression of cyclin D2 increases cyclin D2 protein levels. (A) Schematic of the alleles used to create RIP-Cre;cycD2;ROSA26mT/mG mice (KI mice). Black triangles indicate loxP sites. (B) Representative immunofluorescence staining for insulin or dTomato (red), GFP (green), and DAPI (blue) showing efficient Cre recombinase-mediated recombination in KI β cells. (C) Representative immunofluorescence staining for of cyclin D2 (red) and insulin (green) in WT and KI pancreatic sections. (D) Western blot (left panel) and densitometric quantification of cyclin D2 levels (right panel) in isolated islets from WT and KI mice. Data shown as mean ± SD of 3 independent experiments. ** P<0.01, compared with WT mice.
Overexpression of cyclin D2 promotes β cell self-renewal post-weaning and results in expanded β cell mass
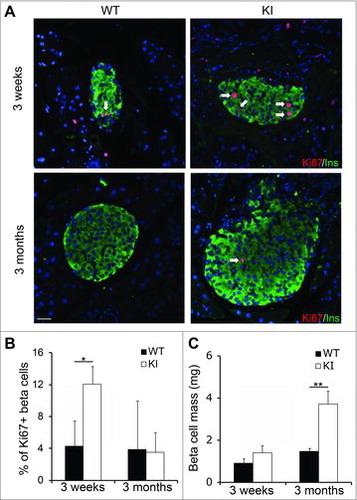
Perinatal pancreatic remodeling includes a brief window of high β cell self-renewal, which declines by weaning at P21. We previously reported that cyclin D2-null (cycD2−/−) mice were born with β cell mass similar to controls at birth, but the loss of cyclin D2 limited β cell self-renewal and resulted in decreased β cell mass as early as 7 d after birth.Citation7 To identify whether cyclin D2 overexpression could enhance postnatal pancreatic β cell self-renewal, we used immunohistochemistry to quantify the proportion of Ki67+ β cells in WT and KI mice. We noted a 2.5-fold increase in the number of Ki67+ β cells in 3-week-old KI mice in comparison to WT, while no difference was seen in the 3 months cohort (). To understand if increased post-weaning self-renewal could result in β cell mass expansion, we measured β cell mass in KI and WT mice at 3 weeks and 3 months of age. At 3 weeks of age, there was a minor increase in β cell mass in the KI pancreas when compared with WT littermates, which was not statistically significant (). This suggests that overexpression of cyclin D2 in the perinatal remodeling period does not drive self-renewal to expand β cell mass. Quantification of β cell mass at 3 months revealed a more than 2-fold increase of β cell mass in KI mice compared with WT mice (). Next, we wanted to investigate if overexpression of cyclin D2 increased β cell survival. We quantified β cell apoptosis by TUNEL assay. Rare TUNEL positive β cells were identified, and the ratio of TUNEL+ β cells were comparable between KI and WT β cells at both 3 weeks and 3 months (data not shown). Taken together, this data suggests that while overexpression of cyclin D2 during the perinatal remodeling period does not increase β cell mass, it may be effective in driving β cell self-renewal to expand β cell mass post-weaning.
Figure 2. Overexpression of cyclin D2 increases β cell proliferation and β cell mass. (A) Representative immunofluorescence staining for insulin (green), Ki67 (red), and DAPI (blue). White arrows indicate Ins+Ki67+ cells. (B) Quantification of the percentage of β cells expressing Ki67 in WT and KI mice at 3 weeks and 3 months of age. (C) Quantification of β cell mass in WT and KI mice at 3 weeks and 3 months of age. Data shown as mean ± SD (n = 3–4 mice per group). *P < 0.05, **P < 0.01.
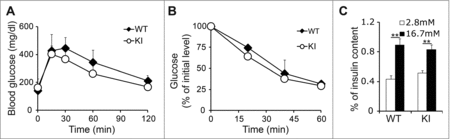
To define whether cyclin D2-mediated islet hyperplasia affected glucose metabolism, we measured glucose tolerance, insulin tolerance, and insulin secretion in 3-month-old KI and WT mice. KI mice displayed a mild improvement in glucose tolerance coupled with increased insulin sensitivity when compared with WT littermates, but differences in the glucose tolerance test or insulin tolerance test were not statistically significant (). This suggested that while there was an increase in β cell mass, insulin sensitivity was not dysregulated. It has been suggested that increased levels of β cell self-renewal may be indicative of a less mature functional state, and thereby lead to decreases in insulin secretion and β cell function.Citation13 To understand if overexpression of cyclin D2 affected islet function, we measured insulin secretion in response to physiologic concentrations of glucose by glucose stimulated insulin secretion assay. We measured insulin secretion in response to basal and high concentrations of glucose in islets isolated from 3-month old WT and KI mice. We did not detect any differences in response to glucose challenge, suggesting that KI islets were functionally mature (). These results indicate that cyclin D2-overexpressing KI islets are functionally mature and able to maintain normal glucose homeostasis.
Figure 3. Overexpression of cyclin D2 in β cells does not diminish β cell function. (A) Glucose tolerance test was performed in 3-month-old WT and KI mice. (B) Insulin tolerance was performed in 3-month-old WT and KI mice. (C) Glucose stimulated insulin secretion was measured in 3-month-old WT and KI mice. n = 4–5 animals per group. Data shown as mean ± SD of 3 independent experiments. **P < 0.01.
Βeta cell specific overexpression of cyclin D2 extends regenerative capacity into advanced age
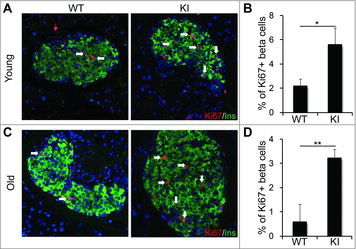
Our previous work has illustrated epigenetic mechanisms that restrict the ability of aged β cells to self-renew in response to metabolic demand for insulin.Citation14-18 Because overexpression of cyclin D2 extended the ability of β cells to self-renew, we investigated if cyclin D2 overexpression could enhance β cell regeneration after β cell injury in aged animals. We used a single dose of streptozotocin (90 mg/kg) to induce β cell death and evaluated the regenerative capacity of KI β cells in young (6 weeks old) and old (8 months old) mice. Immunohistochemical analysis of β cells from young mice showed that KI mice had an increased incidence of large islets when compared with WT mice, and a higher proportion of β cells expressed Ki67 after STZ administration (). Quantification of Ki67+ insulin cells revealed a 2-fold increase in β cell self-renewal in young animals (). In the old mice, immunohistochemical analysis of KI β cells revealed a 3-fold increase in Ki67+ expression in the KI β cells after STZ administration (). While there was a marked increase in self-renewal in the old KI β cells, it is important to note that the proportion of Ki67+ β cells in the old KI group was 50% lower than the young group. This suggests that while overexpression of cyclin D2 does enhance the regenerative capacity of old β cells, age-related restrictions that limit βeta cell self-renewal are sustained. We also measured similar increases in β cell self-renewal and increased β cell mass in old KI animals subjected to a high fat diet (data not shown). Taken together, these data revealed that cyclin D2 overexpression enhances the regenerative capacity of β cells in response to β cell injury.
Figure 4. Overexpression of cyclin D2 enhances β cell replication in old KI mice. (A, B) Representative immunofluorescence staining (A) and quantification (B) for insulin (green), Ki67 (red), and DAPI (blue) in young (6-week-old) WT and KI mice challenged with a single dose of STZ (90 mg/kg). C, D. Representative immunofluorescence staining (C) and quantification (D) for insulin (green), Ki67 (red), and DAPI (blue) in old (8-month-old) WT and KI mice challenged with a single dose of STZ (90 mg/kg). White arrows indicate Ins+Ki67+ cells. Data shown as mean ± SD (n = 3–4 mice per group). *P < 0.05, **P < 0.01.
β cell-specific cyclin D2 re-expression restores self-renewal and the regenerative capacity in the β cells of cyclin D2−/− mice
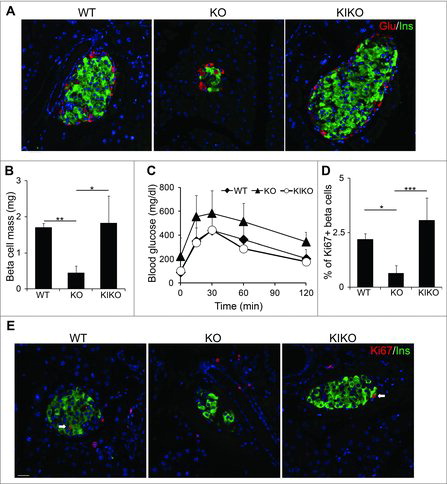
Our previous work concluded that cyclin D2-mediated β cell self-renewal is the primary means of β cell mass expansion both in homeostasis and in response to metabolic demand for insulin.Citation7,10 At the time, these studies were limited by the use of the global cyclin D2−/− model and the inability to reconstitute cyclin D2 expression in cyclin D2−/− β cells. To address these shortcomings, we crossed KI mice with cyclin D2−/− mice to restore cyclin D2 expression in specifically in β cells (referred to herein as KIKO). There were no differences in the body mass and pancreas mass between WT, KI, and KIKO animals (data not shown). We used immunohistochemistry to evaluate islet architecture of the KIKO mouse. When we compared KIKO islets to the both wildtype and cyclin D2−/− islets, we found that KIKO mice displayed similar islets size and β cell mass to WT mice (). We quantified β cell mass in WT, cyclin D2−/−, and KIKO animals at 3 months old. While the cyclin D2−/− β cell mass was considerably decreased in comparison to WT animals, reconstituting cyclin D2 expression in cyclin D2−/− insulin cells resulted in a restoration of β cell mass (). After concluding that reconstituting cycD2 expression can restore β cell mass in cyclin D2−/− animals, we evaluated the ability of KIKO β cells to maintain glucose homeostasis. Consistent with previous findings, cyclin D2−/− mice were glucose intolerant, but reconstituting cyclin D2 expression in the KIKO mice completely restored glucose clearance comparable to WT littermates ().Citation7,8,10 We next examined if reconstitution of cyclin D2 expression could enhance β cell regeneration in the cyclin D2−/− pancreas after STZ injury. We administered a single dose of STZ (90 mg/kg) to induce partial β cell loss and measured β cell regeneration by quantification of Ki67+ β cells after 7 d. While the cyclin D2−/− mice showed a 4-fold decrease in β cell regeneration, reconstitution of cyclin D2 expression in β cells restored β cell regeneration in the KIKO mice (). Taken together, these experiments suggest that overexpression of cyclin D2 specifically in the β cell compartment is sufficient to restore β cell function, mass, and regenerative capacity in the cyclin D2−/− background.
Figure 5. Reconstituting cyclin D2 expression restores β cell mass, function, and regenerative capacity. (A) Representative immunofluorescence staining glucagon (red), insulin (green) and DAPI (blue) in 6-week-old WT, KO, and KIKO mice. (B) Quantification of β cell mass in 6-week-old WT, KO, and KIKO mice. (C) GTT was performed in 3-month-old WT, KO, and KIKO mice. (D, E) Representative immunofluorescence staining (F) and quantification (E) for insulin (green), Ki67 (red), and DAPI (blue) in 6-week-old WT, KO and KIKO mice treated with a single dose of STZ (90 mg/kg). White arrows indicate Ins+Ki67+ cells. Data shown as mean ± SD (n = 3 mice per group). * P < 0.05, **P < 0.01, ***P < 0.005.
Discussion
An adequate and functional β cell mass is required to maintain glucose homeostasis. Increasing numbers of patients in need of insulin replacement therapy is driving a demand to develop novel therapeutic strategies to expand functional β cell mass. Understanding the mechanisms that drive β cell self-renewal is the subject of intense investigation. Our work and others have shown that cyclin D2 is required for postnatal β cell self-renewal and under conditions of insulin resistance.Citation7,8,10 Elevated cyclin D2 expression and enhanced β cell growth have been observed in mouse models such as high fat diet treatment,Citation19 partial pancreatectomy,Citation12 glucose infusion,Citation20 or exposure to prolactin and growth hormone,Citation21 indicating that increased cyclin D2 may be a necessary component of the cell cycle machinery that mediates β cell reentry into cell cycle.
In this study, we sought to address if cyclin D2 was sufficient to drive β cell expansion through self-renewal. Our data indicates that while a 2-fold increase in the expression of cyclin D2 does not influence β cell mass expansion during the early perinatal remodeling period, it increased β cell self-renewal post-weaning, thereby expanding β cell mass in adulthood. The phenotype of KI mice resembles the Cdk4R24C transgenic mice, which expressed a constitutively active form of the cyclin D2 binding partner, Cdk4. Cdk4R24C mice displayed pancreatic hyperplasia due to abnormal proliferation of β cells by 3 months of age.Citation22 Conversely, our results were very different from the transgenic cyclin D2 T280A β cells, where it was reported that accumulation of the stabilized form of cyclin D2 did not increase β cell proliferation, but decreased β cell apoptosis and increased β cell survival, resulting in increased β cell mass and eventual tumorigenesis.Citation23 Our KI mice did not exhibit signs of tumorigenesis by 8 months of age, suggesting that the endogenous mechanisms that regulate cyclin D2 turnover are sufficient to control for cell cycle-dependent cyclin D2-mediated overgrowth of β cell mass.
Elegant experiments quantifying CldU and IdU thymidine analogs incorporation into β cells illustrated that β cell self-renewal is restricted by a refractory period that slows self-renewal under basal conditions. This refractory period is not static, as the authors demonstrated that it could be foreshortened in response to partial pancreatectomy.Citation24 In our KI model, β cell self-renewal was enhanced in aged KI mice treated with STZ. It is possible that sustained expression of cyclin D2 may foreshorten the replication refractory period to promote β cell self-renewal and facilitate regeneration after β cell injury. The mechanisms that regulate the timing of the refractory period are not entirely clear, but may be dependent on the balance between pro-proliferative activity of cyclin D2 and cdk4 complexes and anti-proliferative inhibition by p16ink4a.
We and others have previously reported that there is an age-dependent decline in β cell self-renewal through downregulation of Bmi1, upregulation of Ezh2, and subsequent accumulation of p16Ink4a.Citation16-18 Neither consumption of a high fat diet, injury by STZ, nor short-term exposure to GLP1 stimulated increase β cell self-renewal in old mice due to a loss of Ezh2-mediated repression of p16ink4a.Citation14,25 Subsequent studies revealed that overexpression of Ezh2 was insufficient to repress p16ink4a expression in old mice, but could be used in combination with other epigenetic modulators to re-ignite β cell replication in old animals.Citation18 The KI mouse model seems to bypass this epigenetic restriction of β cell self-renewal by overwhelming the stoichiometric balance between cyclin D2 pro-proliferative signals and p16Ink4a anti-proliferative signals to drive β cell self-renewal in adulthood.
In the absence of cyclin D2 pro-proliferative signaling, β cells are unable to self-renew to meet metabolic demand for insulin. In this study, we determined that expression of cyclin D2 exclusively in β cells was sufficient to drive postnatal β cell self-renewal to establish a functional β cell mass able to maintain glucose homeostasis. Interestingly, reconstitution of cyclin D2 expression in the cyclin D2−/− mouse did not expand β cell mass above that of wildtype littermates (). This could be interpreted to mean that the expression of cyclin D2 in other pancreatic compartments may have a minor contribution to establishing postnatal β cell mass, which is consistent with other studies in pancreatic injury models.Citation11,26
Our results suggest that cyclin D2 overexpression has the potential to enhance β cell regeneration. Previous studies using human islets have concluded overexpression of cdk6 or increasing signals upstream of cyclin D2 in human islets results in the accumulation of D-type cyclins and re-entry into the cell cycle.Citation27,28 Taken together, this suggests cyclin D2, or a complementary cyclin/cdk analog in humans, is a potential target that can manipulated to promote β cell expansion for the treatment of diabetes.
Research design and methods
Mouse husbandry
Stop-floxed-Cyclin D2 conditional KI mice that harbor a mouse cyclin D2 allele encoding mouse cyclin D2 targeted immediately after a loxP-flanked transcriptional stop sequence at the ROSA26R locus were generated at Indiana University in the DBA/2J background. Targeted disruption of the cyclin D2 allele and the RIP-Cre mice have been described previously and were on a C57BL/6 background.Citation7,29 The Rosa26R-mTmG (JAX 007576) line was obtained from Jackson Laboratory in the 129Sv/J background. All mice used in these experiments were a mixture of the given backgrounds and were littermates from the mixture. Mice were kept under a 12-h light/dark circle with standard diet. All animal protocols were approved by the Chancellor's Animal Research Committee at the University of California, Los Angeles.
Western blotting
Islet isolation was performed as described previously.Citation14 Lysates extracted by tissue extraction buffer (Invitrogen) were resolved by SDS-PAGE, followed by transferring to polyvinylidene fluoride membrane for immunoblotting. The membranes were probed with specific antibodies against cyclin D2 (Santa Cruz), and β-tubulin (Sigma-Aldrich). For densitometric analysis, protein levels were normalized to the protein levels of housekeeping gene β-tubulin. The data presented is representative of 3 independent experiments, using islet lysate from different mice in each independent experiment.
Immunohistochemistry
Immunohistochemistry was performed as described previously.Citation14 Antibodies used were: guinea pig anti-insulin (1:400; Dako), mouse anti-Ki67 (1:40; BD Pharmingen), chicken anti-GFP (1:250; Aves Labs), rabbit anti-cyclin D2(1:3000; Santa Cruz) antibody and fluorescein- isothiocyanate- or Cy3-conjugated secondary antibodies (The Jackson Laboratory). After mounting with Vectashield (Vector Laboratories), all the slides were viewed using a Leica DXMRA microscope and images acquired using Openlab software. The percentage of Ki67+ β cells was determined by counting how many insulin+ β cells were also Ki67+. A minimum of 1500 β cells from 3 different sections were counted for each animal. The data are expressed as the average percentage of Ki67+ β cells per genotype, n = 3 animals per genotype.
β cell mass
β cell mass was measured as described previously.Citation10 In brief, 4 to 6 sections from each pancreas were stained with anti-insulin antibody and scanned by a Leica DM6000 microscope. Montage images were made by ImageJ software. The cross-sectional areas of pancreas and β cells were determined by ImagePro software. β cell mass per pancreas was estimated as the product of the relative cross-sectional area of β cells per total tissue and the weight of the pancreas and calculated by examining pancreata from at least 3 animals for each genotype.
STZ administration
A single dose of 90 mg/kg STZ (Sigma-Aldrich) in citrate buffer (pH 4.5) was injected intraperitoneally, and pancreata were harvested for proliferation index analysis after 7 d as described previously.Citation14
Metabolic analysis
Glucose tolerance testing was performed after overnight fasting, and blood glucose levels were measured before intraperitoneal injection of glucose (2 mg dextrose/g body wt) and 15, 30, 60, and 120 min after injection. Glucose stimulated insulin secretion experiment was performed after 1 hour equilibration in Kreb's buffer containing 2.8 mM glucose. Islets were sequentially incubated in Kreb's buffer containing 2.8 mM Glucose and 16.7 mM glucose for 1 hour, respectively. Supernatant was collected to measure insulin secretion and islets were harvested to lyse in acid ethanol to extract insulin. Insulin was measured with mouse insulin ELISA kit (Mercodia).
Statistical analysis
All data were summarized as the means ± SD. Mean and SD values were calculated from at least triplicates of representative experiments. Comparisons among 2 groups were made with a 2-tailed paired student's t-test at a p<0.05 significance level. Comparisons among the 3 groups were made with 1-way Analysis of Variance (ANOVA), with 2 Bonferroni-adjusted post hoc pairwise comparisons between KO and the other 2 groups. Serial blood glucose levels from GTT were analyzed with repeated measures ANOVA, with Dunnet test to compare overall glucose levels in KO to WT and KIKO groups. Statistical analysis was performed using SAS/STAT© v9.2 software at a p <0.05 significance level.
Abbrevations
| Glu | = | glucagon |
| GSIS | = | glucose stimulated insulin secretion |
| GTT | = | glucose tolerance testing |
| Ins | = | insulin |
| ITT | = | insulin tolerance testing |
| KI | = | knock-in |
| KO | = | knockout |
| WT | = | wild type. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965; 14(10):619-33; PMID:5318831; https://doi.org/10.2337/diab.14.10.619
- Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes 1978; 27(Suppl 1):251-61; PMID:75815; https://doi.org/10.2337/diab.27.1.S251
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52(1):102-10; PMID:12502499; https://doi.org/10.2337/diabetes.52.1.102
- Meier JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia 2008; 51(5):703-13; PMID:18317728; https://doi.org/10.1007/s00125-008-0936-9
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429(6987):41-6; PMID:15129273; https://doi.org/10.1038/nature02520
- Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012; 97(9):3197-206; PMID:22745242; https://doi.org/10.1210/jc.2012-1206
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 2004; 114(7):963-8; PMID:15467835; https://doi.org/10.1172/JCI22098
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 2005; 25(9):3752-62; PMID:15831479; https://doi.org/10.1128/MCB.25.9.3752-3762.2005
- Kushner JA. Beta-cell growth: an unusual paradigm of organogenesis that is cyclin D2/Cdk4 dependent. Cell Cycle 2006; 5(3):234-7; PMID:16410729; https://doi.org/10.4161/cc.5.3.2399
- Georgia S, Hinault C, Kawamori D, Hu J, Meyer J, Kanji M, Bhushan A, Kulkarni RN. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes 2010; 59(4):987-96; PMID:20103709; https://doi.org/10.2337/db09-0838
- Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008; 132(2):197-207; PMID:18243096; https://doi.org/10.1016/j.cell.2007.12.015
- He LM, Sartori DJ, Teta M, Opare-Addo LM, Rankin MM, Long SY, Diehl JA, Kushner JA. Cyclin D2 protein stability is regulated in pancreatic beta-cells. Mol Endocrinol 2009; 23(11):1865-75; PMID:19628581; https://doi.org/10.1210/me.2009-0057
- Szabat M, Lynn FC, Hoffman BG, Kieffer TJ, Allan DW, Johnson JD. Maintenance of β-Cell Maturity and Plasticity in the Adult Pancreas. Dev Biol Concepts Adult Physiol 2012; 61(6):1365-71
- Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009; 58(6):1312-20; PMID:19228811; https://doi.org/10.2337/db08-1651
- Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009; 58(6):1365-72; PMID:19265026; https://doi.org/10.2337/db08-1198
- Chen H, Gu X, Su Ih, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 2009; 23(8):975-85; https://doi.org/10.1101/gad.1742509
- Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev 2009; 23(8):906-11; PMID:19390085; https://doi.org/10.1101/gad.1742609
- Zhou JX, Dhawan S, Fu H, Snyder E, Bottino R, Kundu S, Kim SK, Bhushan A. Combined modulation of polycomb and trithorax genes rejuvenates β cell replication. J Clin Invest 2013; 123(11):4849-58; PMID:24216481; https://doi.org/10.1172/JCI69468
- Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive beta-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab 2013; 305(1):E149-59; PMID:23673159; https://doi.org/10.1152/ajpendo.00040.2013
- Alonso LC, Yokoe T, Zhang P, Scott DK, Kim SK, O'Donnell CP, Garcia-Ocana A. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 2007; 56(7):1792-801; PMID:17400928; https://doi.org/10.2337/db06-1513
- Friedrichsen BN, Richter HE, Hansen JA, Rhodes CJ, Nielsen JH, Billestrup N, Moldrup A. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol Endocrinol 2003; 17(5):945-58; PMID:12586844; https://doi.org/10.1210/me.2002-0356
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999; 22(1):44-52; PMID:10319860; https://doi.org/10.1038/8751
- He LM, Sartori DJ, Teta M, Opare-Addo LM, Rankin MM, Long SY, Diehl JA, Kushner JA. Cyclin D2 Protein Stability Is Regulated in Pancreatic β-Cells. Mol Endocrinol 2009; 23(11):1865-75; PMID:19628581; https://doi.org/10.1210/me.2009-0057
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007; 12(5):817-26; PMID:17488631; https://doi.org/10.1016/j.devcel.2007.04.011
- Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009; 58(6):1312-20. PMID:19228811; https//doi.org/10.2337/db08-1651
- Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, et al. Adult Duct-Lining Cells Can Reprogram into beta-like Cells Able to Counter Repeated Cycles of Toxin-Induced Diabetes. Dev Cell 2013; 26(1):86-100; PMID:23810513; https://doi.org/10.1016/j.devcel.2013.05.018
- Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, Cherok E, Takane KK, Scott DK, Stewart AF. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010; 59(8):1926-36; PMID:20668294; https://doi.org/10.2337/db09-1776
- Chen H, Kleinberger JW, Takane KK, Salim F, Fiaschi-Taesch N, Pappas K, Parsons R, Jiang J, Zhang Y, Liu H, et al. Augmented Stat5 Signaling Bypasses Multiple Impediments to Lactogen-Mediated Proliferation in Human beta-Cells. Diabetes 2015; 64(11):3784-97; PMID:26159175; https://doi.org/10.2337/db15-0083
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000; 127(11):2317-22; PMID:10804174