
ABSTRACT
Oxidative stress is considered as an important pathogenic factor in many human diseases including Fanconi anemia (FA), an inherited bone marrow failure syndrome with extremely high risk of leukemic transformation. Members of the FA protein family are involved in DNA damage and other cellular stress responses. Loss of FA proteins renders cells hypersensitive to oxidative stress and cancer transformation. However, how FA cells respond to oxidative DNA damage remains unclear. By using an in vivo stress-response mouse strain expressing the Gadd45β-luciferase transgene, we show here that haematopoietic stem and progenitor cells (HSPCs) from mice deficient for the FA gene Fanca or Fancc persistently responded to oxidative stress. Mechanistically, we demonstrated that accumulation of unrepaired DNA damage, particularly in oxidative damage-sensitive genes, was responsible for the long-lasting response in FA HSPCs. Furthermore, genetic correction of Fanca deficiency almost completely abolished the persistent oxidative stress-induced G2/M arrest and DNA damage response in vivo. Our study suggests that FA pathway is an integral part of a versatile cellular mechanism by which HSPCs respond to oxidative stress.
Introduction
Fanconi anemia (FA) is a rare inherited disease with 21 complementation groups identified thus far.Citation1-Citation10 Mutations in any of the FA genes (FANCA-V) leads to clinical manifestations characterized by developmental abnormalities, progressive bone marrow failure (BMF) and a high risk of developing cancer, especially leukemia.Citation1,Citation6,Citation10 At the cellular level, FA is characterized by chromosomal instability and cross-linker sensitivity, which serves as a clinical diagnostic hallmark of FA.Citation1,Citation6 The prominent role of the FA protein family has been implicated in the network of DNA damage response (DDR), which includes DNA repair pathways such as homologous recombination (HR) and non-homologous end-joining (NHEJ).Citation3,Citation5 In response to DNA damage or DNA replication stress, 8 of FA proteins (namely, FANCA, B, C, E, F, G, L and M) form a nuclear complex, which functions as an ubiquitin ligase. This ubiquitin ligase monoubiquitinates 2 downstream FA proteins, FANCD2 and FANCI, which then recruit other downstream FA proteins and possibly other DNA repair factors to nuclear loci containing damaged DNA and consequently influence important cellular processes, such as DNA replication, cell cycle control, and DNA damage repair.Citation11,Citation12
One common type of DNA damage in vivo is caused by oxidative stress, defined by an imbalance between the production of reactive oxygen species (ROS) and cellular antioxidant defense. Oxidative stress can cause oxidative damage to biomolecules, such as lipids, proteins and DNA,Citation13,Citation14 and has been considered as an important pathogenic factor in leukemia-prone bone marrow diseases, including FA.Citation5,Citation15 Cellular responses inducing resistance to oxidative stress are important for cellular survival, organism lifespan, and cancer prevention.Citation15 However, mammalian factors regulating resistance to oxidative stress have not been well characterized.Citation15
It is known that loss of FA proteins renders cells a high predisposition to cancer transformation.Citation1,Citation5,Citation6,Citation10,Citation16,Citation17 An intensive body of evidence suggests that FA proteins play crucial role in oxidative stress signaling in variety of cell types including HSPCs.Citation8,Citation15,Citation18,Citation19 Haematopoietic cells are exposed to various ROS even under physiologic condition, which induce a variety of responses such as cellular proliferation and growth inhibition.Citation20-Citation23 It has been postulated that excessive apoptosis of haematopoietic stem/progenitor cells (HSPCs) induced by oxidative stress is a critical factor in the pathogenesis of BMF and leukemia progression in FA.Citation5,Citation24,Citation25 In fact, FA has been proposed as a unique disease model characterized by abnormal accumulation of ROS and dysfunctional response to oxidative stress.Citation15,Citation19 However, how FA HSPCs respond to oxidative DNA damage remains largely unknown.
In the present study, we used an in vivo stress-response model expressing the luciferase transgene under the control of the promoter of the stress-responsive gene Gadd45β and showed that HSPCs from mice deficient for the Fanca or Fancc gene persistently responded to oxidative stress. Significantly, genetic correction of Fanca deficiency shortens the long-lasting oxidative stress-induced response in FA HSPCs. Mechanistically, we demonstrated that accumulation of unrepaired DNA damage, particularly in oxidative damage-sensitive genes, was responsible for the long-lasting response in FA HSPCs.
Results
FA HSPCs are hypersensitive to oxidative stress
To study the role of FA proteins in response to oxidative stress specifically in HSPCs, we first treated bone marrow (BM) LSK (Lin−Sca1+c-kit+) cells from Fanca−/−, Fancc−/− and their wild-type (WT) littermates with increasing doses of paraquat, a chemical that can induce accumulation of reactive oxygen species (ROS).Citation26 We found that paraquat induces apoptosis in both WT and Fanca−/− or Fancc−/− LSK cells (Fig S1A) in a dose-dependent manner as detected by Annexin V/7AAD staining (, S1B). However, we noted that Fanca−/− or Fancc−/− cells were much more sensitive to paraquat treatment than their WT controls. Furthermore, paraquat also induced a dose-dependent decrease in colony formation of the HSPC compartment, as determined by CFU assay, with Fanca−/− or Fancc−/− HSPCs showing more profound effect than their WT counterparts (, S1C).
Figure 1. FA cells are hypersensitive to oxidative stress. (A) FA haematopoietic stem progenitor cells (HSPCs) are more sensitive to oxidative stress. Bone marrow LSK cells (Lin−Sca1+c-kit+ cells) isolated from Fanca−/− mice or their wild-type (WT) littermates were treated with different doses of paraquat for 24 hours followed by Flow cytometric analysis for apoptosis using Annexin V and 7AAD staining. Cells were gated on LSK population. Representative images (left) and quantification (right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group). (B) Oxidative stress reduces colony formation capacity of FA stem progenitor cells. LSK cells from mice described in (A) were treated with different doses of paraquat for 2 hours then plated in cytokine-supplemented methycellulose medium. Colonies were enumerated on day 7 after plating. The error bars represent the standard deviation from 3 independent experiments (n = 6 per group). (C) Oxidative stress compromises FA HSPC function in vivo. 1,000 paraquat treated WT or Fanca−/− LSK cells along with 3 × 105 BM cells from congenic BoyJ mice were transplanted into lethally irradiated BoyJ recipients. Donor-derived total CD45.2+ cells (Left), progenitors (LSK cells, Middle) and stem cells (SLAM: Lin−Sca1+c-kit+CD48+CD150− cells, Right) were assessed by Flow cytometry at 4 month post-transplant. Quantifications (Right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group).
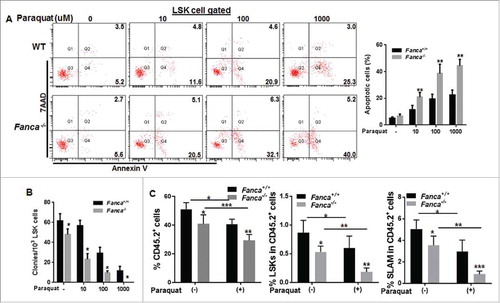
To confirm the observation in vivo, we performed bone marrow transplantation (BMT) by injecting paraquat treated or untreated LSK cells from WT mice or Fanca−/− mice along with 3 × 105 BM cells from congenic BoyJ mice into lethally irradiated BoyJ recipients.Citation11 Flow cytometry analysis at 4-month post-transplant revealed that oxidative stress-induced by paraquat significantly reduces donor-derived hematopoiesis in both WT and Fanca−/− cells (). However, the reduction was much profound in Fanca−/− cells transplanted recipients than that in WT cells transplanted BoyJ recipients, especially in the phenotypic (LSKCD150+CD48−, SLAM)Citation27 HSC compartment (). Taken together, these results indicate that Fanca−/− and Fancc−/− HSPCs are hypersensitive to oxidative stress.
FA HSPCs exhibit prolonged response to oxidative stress
To determine the kinetics of HSPC response to oxidative stress, we assessed oxidative stress-induced G2/M cell cycle arrest, which is a hallmark of FA cells.Citation28 LSK cells were treated with 100 μM of paraquat for 2 hours followed by drug-free culture for up to 72 hours. We found that Paraquat-induced G2/M arrest peaked at 48 hours in WT LSK cells and returned to cycle at 72 hours after released from the paraquat treatment (, S2A). Strikingly, a high percentage of Fanca−/− or Fancc−/− LSK cells remained arrested in G2/M phase throughout the 72-hour culture after the release (, S2A). This suggests a long-lasting response of FA HSPCs to oxidative stress in vitro.
Figure 2. FA cells exhibit persistent response to oxidative stress in vitro and in vivo. (A) Paraquat treatment induces G2/M arrest in FA HSCs. Low density bone marrow cells (LDBMCs) isolated from Fanca−/− mice or their wild-type (WT) littermates were treated with 100 μM paraquat for 2 hours followed by drug-free culture in fresh medium for the indicated time intervals. The cells were then subjected to cell cycle analysis by PtdIns staining. Cells were gated on LSK population. Representative flow plots (left) and quantification (right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group). (B) Paraquat induces prolonged DNA damage in FA cells in vivo. FACS-sorted BM LSK cells isolated from Luc-Fanca−/− or their WT littermates along with 3 × 105 BM cells from congenic BoyJ mice were transplanted into lethally irradiated recipients. Four months later, the recipients with similar donor chimerism were treated with a single dose of paraquat (10mg/kg) followed by imaging luciferase expression at different time points. Luciferase intensity was measured by IVIS system at indicated time point after injection. Luminescence scale is in p/s/cm2/sr.
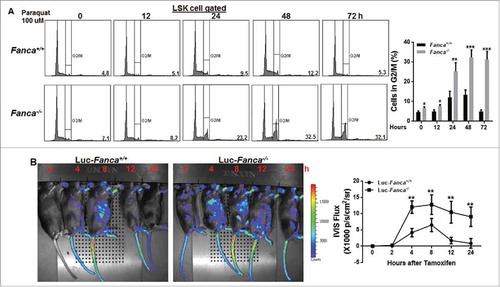
To determine the dynamic response of HSPCs to oxidative stress in vivo, we crossed our FA mice to the Luc-Gadd45β reporter mice, which express the luciferase transgene under the control of the promoter of the stress-responsive gene Gadd45βCitation29 and allow for non-invasive in vivo imaging stress-induced expression of the luciferase marker.Citation11,Citation30 FACS-sorted BM LSK cells isolated from Luc-Fanca−/− , Luc-Fancc−/− mice or their WT littermates were transplanted into lethally irradiated recipients. Four months later, the recipients with similar donor chimerism (Fig S2B) were treated with a single dose of paraquat followed by non-invasive imaging luciferase expression at different time points. This experimental design allowed us to examine paraquat-induced oxidative stress response specifically in donor-derived HSPCs and their progeny in the recipients.Citation11,Citation30 We observed a persistent luciferase expression in recipients transplanted with Luc-Fanca−/− or Luc-Fancc−/− LSK cells during the 24-hour period post-paraquat injection (, S2C). In contrast, recipients of Luc-WT LSK cells showed transient paraquat-induced luciferase expression with a peak at 8 hours and returned to the basal level at 12 hours post-injection (, S2C). These results indicate that FA HSPCs exhibit persistent response to oxidative stress in vivo.
Unrepaired DNA damage is responsible for persistent response to oxidative stress in FA HSPCs
To explore the mechanism underlying persistent oxidative stress response, we examined the repair kinetics of paraquat-induced DNA damage by immunoblotting for γ-H2AX, a robust indicator of DNA strand breaks.Citation31 We treated freshly isolated LSK cells from WT, Fanca−/− or Fancc−/− mice with paraquat for 2 hours then released them for the indicated time intervals followed by monitoring the kinetics of γ-H2AX clearance at different time points. The expression of γ-H2AX was evident at 4 hours after paraquat treatment in both WT and Fanca−/− or Fancc−/− LSK cells (, S3A). However, the repair kinetics showed strikingly different between these 2 genotypes. Specifically, WT LSK achieved a complete γ-H2AX clearance within 12 hours after release from paraquat treatment; while in Fanca−/− or Fancc−/− LSK cells, high levels of γ-H2AX expression was sustained for 48 hours after release into the fresh medium (, S3A). Consistently, deletion of Fanca or Fancc gene in LSK cells led to a persistent and high percentage of cells containing DNA migration tail throughout the 48-hour post-paraquat period as determined by Comet assay (, S3B). In contrast, WT LSKs exhibited a bell-like kinetics of DNA damage clearance with the paraquat-induced comet tail peaking at 24 hours and returning to basal level at 48 hours (, S3B). These results indicate a persistent accumulation of unrepaired DNA damage in FA HSPCs.
Figure 3. Unrepaired DNA damage is responsible for persistence of FA cells to oxidative stress. (A) Oxidative stress leads to persistent γ-H2AX expression in FA LSK cells in vitro. LSK cells from Fanca−/− mice or their WT littermates were treated with 100 μM paraquat for 2 hours and released for the indicated time intervals. Whole cell lysates were then extracted for immunoblot analysis using antibodies against γ-H2AX or β-actin. (B) Oxidative stress induces persistent DNA damage in FA HSCs. Cells described in (A) were subjected to comet assay and a total of 300 cells were counted under microscope for DNA migration tail. Representative microscope images (Left) and quantification (Right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group). (C) Oxidative stress induces persistent damage to oxidative damage-sensitive gene promoters. Genomic DNA from cells described in (A) were subjected to 8-oxo-dG incorporation assay. Untreated cells were used as control. Shown is the fold increase in 8-oxoguanie (8-oxo-dG) incorporation into promoters of Calmodulin 1 (CaM1) relative to those of GAPDH. (D) Increased 8-oxodG accumulation in the promoters of antioxidant genes in FA HSCs. Cells described in (B) were treated with 100 μM of Paraquat for 2 hours and released in the fresh medium for the indicated time intervals followed by ChIP using an antibody against 8-oxodG. Precipitated samples were then subjected to PCR using primers specific for promoter regions of CaM1 gene. Representative images (left) and quantifications (right) are shown. The intensities of DNA bands were quantified using ImageJ software (NIH). Results are means ± SD of 3 independent experiments. (E) Oxidative stress induces persistent DNA damage in FA mice. Fanca+/+ or Fanca−/− mice were i.p. injected with single dose of paraquat followed by bone marrow cells (BMCs) isolation at different time points. The cells were then subjected to Flow cytometric analysis for γ-H2AX. LSK compartment were gated. Representative flow plots (left) and quantification (right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group).
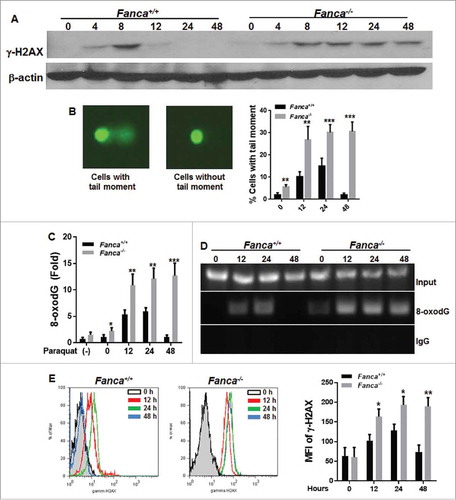
Because oxidative stress can induce a significant increase in oxidative DNA damage in the promoter regions of certain genes,Citation18,Citation32 we next determined whether loss of FA proteins also led to a persistent accumulation of oxidative DNA damage in oxidative damage-sensitive gene promoters. Using the well-established 8-oxo-dG incorporation assay for oxidative DNA damage,Citation32 we observed persistent 8-oxo-dG incorporation into the promoter region of Calmodulin 1 (CaM1), an oxidative stress-sensitive gene,Citation32,Citation33 in Fanca−/− or Fancc−/− LSK cells, which peaked at 12 hours and remained high up to 48 hours after paraquat treatment (, S3C). In contrast, WT LSK cells exhibited the typical bell-shape repair kinetics with a peaked 8-oxo-dG incorporation at 12–24 hours and then declined to basal level at 48 hours after paraquat (, S3C). It is noteworthy that even under steady-state, Fanca−/− or Fancc−/− LSK cells exhibited increased 8-oxo-dG incorporation in the promoter region of CaM1 compared with WT cells, albeit not statistically significant. We also performed chromatin immunoprecipitation (ChIP) analysis of the CaM1 gene promoter with an antibody against 8-oxo-dG. Again, we observed a prolonged accumulation of 8-oxo-dG in the promoter of CaM1 in Fanca−/− and Fancc−/− LSK cells, while in WT LSK cells, the level of 8-oxo-dG appeared to peak at 24 hours after paraquat treatment and declined to background level at 48 hours after the treatment (, S3D).
To confirm these findings in vivo, we analyzed γ-H2AX levels by flow cytometry in the LSK compartment of mice treated with paraquat and killed at different time points. The results were generally consistent with the in vitro observations described above. Specifically, the γ-H2AX level in LSK cells from WT littermates peaked at 24 hours but was reduced to baseline at 48 hours after paraquat injection (and S3E). However, LSK cells from Fanca−/− and Fancc−/− mice exhibited persistently high levels of γ-H2AX during the period of 12–48 hours post-injection ( and S3E). Collectively, these results support the notion that accumulation of unrepaired DNA damage may be responsible for the observed long-lasting response to oxidative stress in the FA HSPCs.
Genetic correction of FA deficiency prevents prolonged response to oxidative stress
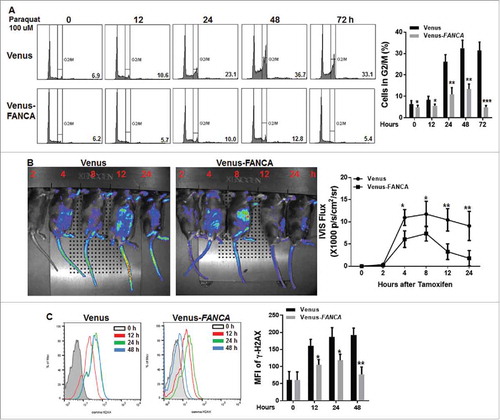
Next, we performed rescue experiments to determine whether genetic correction of Fanca deficiency could shorten the prolonged response of Fanca−/− HSPCs to oxidative stress. To this end, we ectopically expressed a full-length FANCA cDNA in BM LSK cellsCitation30 isolated from Luc-Fanca−/− mice and determined cell cycle status of the transduced cells after paraquat treatment. The results showed that complementation of Fanca−/− HSPCs with the FANCA cDNA greatly shortened the long-lasting paraquat-induced G2/M arrest in vitro with a peak at 48 hours after paraquat treatment and return to cell at 72 hours post treatment (). We also transplanted the transduced cells into lethally irradiated recipients and determined the in vivo response to oxidative stress by measuring the expression of Gadd45β-controlled luciferase reporter. As shown in , luciferase level declines significantly faster in the recipients transplanted with the FANCA-expressing HSPCs compared with those carrying vector control, which peaked at 8 hours but was reduced to baseline at 12 hours after paraquat injection. Accordingly, the γ-H2AX level in LSK cells from the FANCA-expressing lentivirus-transduced HSPCs exhibited a complete γ-H2AX clearance within 48 hours after release from paraquat treatment (), suggesting that genetic correction of Fanca deficiency almost completely rescues oxidative stress-induced long-lasting response in FA HSPCs.
Figure 4. Genetic correction of FA deficiency prevents prolonged response to oxidative stress. (A) Complementation Fanca−/− HSPCs with human FANCA abolishes paraquat-induced G2/M arrest. LDBMCs isolated from Fanca−/− mice or their WT littermates were transduced with lentiviral vector expressing Venus or Venus-FANCA. FACS-sorted Venus+ cells were then treated with 100 μM paraquat for 2 hours followed by drug-free culture in fresh medium for the indicated time intervals. The cells were then subjected to cell cycle analysis by PtdIns staining. Cells were gated on LSK population. Representative Flow plots (Left) and quantification (right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 4 per group). (B) Genetic correction of Fanca deficiency in HSPCs shortens oxidative stress-induced persistent response in vivo. LSK cells isolated from Luc-Fanca+/+ mice or their WT littermates were transduced with lentiviral vector expressing Venus or Venus-FANCA. Sorted Venus+ cells were then transplanted into lethally irradiated recipients. Four months later, the recipients with similar donor chimerism were treated with a single dose of paraquat (10mg/kg) followed by imaging luciferase expression at different time points. Luciferase intensity was measured by IVIS system at indicated time point after injection. Representative Flow plots (Left) and quantifications (Right) are shown. Luminescence scale is in p/s/cm2/sr. (C) Gene correction of Fanca deficiency rescues oxidative stress induced long-lasting γ-H2AX expression in FA LSK cells in vivo. Recipients described in (B) were i.p injected with a single dose of paraquat followed by BMCs isolation at different time points. The cells were then subjected to Flow cytometric analysis for γ-H2AX. LSK compartment were gated. Histogram (Left) and quantification (right) are shown. Results are means ± standard deviation (SD) of 3 independent experiments (n = 6 per group).
Discussion
Cells from patients with FA gene mutations have high predisposition to leukemia and other cancers.Citation1,Citation6,Citation10 Little is known about whether these mutant cells have high susceptibility to oxidative DNA damage and cancer transformation. In the present study, we investigated the response of FA HSPCs to oxidative stress and found that the FA pathway plays critical roles in response to oxidative stress. Our study identifies a role for FA proteins in safeguarding the cellular antioxidant defense system, which links the FA pathway with the oxidative stress response. There are several findings that highlight the significance of our study: 1) FA HSPCs are hypersensitive to oxidative stress; 2) FA HSPCs exhibit persistent response to oxidative DNA damage; 3) Accumulation of unrepaired oxidative DNA damage is identified as one mechanism responsible for the long-lasting response to oxidative DNA damage in FA HSPCs; 4) Genetic correction of Fanca deficiency abolishes the prolonged response of FA HSPCs to oxidative stress.
We observed increased apoptosis and reduced colony formation capacity of FA HSPCs in response to paraqaut treatment. These results are consistent with recently documented FA oxidant hypersensitivity using primary and immortalized cell cultures as well as patient samples. For example, Fancc−/− mouse embryonic fibroblasts (MEFs) have been shown hypersensitive to H2O2 through overactivation of apoptosis signal-regulating kinase 1 (ASK) and pretreatment with antioxidants NAC enhances MEF cell survival.Citation34 Mice with combined deficiencies of the antioxidative enzyme, Cu/Zn superoxide dismutase, and Fancc genes (Fancc−/− Sod−/− mice) demonstrated a defective hematopoiesis.Citation35 Although these observations point to the involvement of FA proteins in oxidative stress response, the molecular mechanism by which FA proteins function to modulate oxidative stress response has not been defined.
At the molecular level, 3 components of FA core complex, FANCA, FANCC, FANCG have been shown associated with factors involved in redox signaling.Citation15, Citation34-Citation39 We previously showed that the FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoter in human cell lines.Citation18 Here we show that prolonged accumulation of DNA damage in the oxidative sensitive-gene promoters, due to a lagging repair kinetics (), is one mechanism responsible for the persistent oxidative stress response observed in FA HSPCs. Our results appear to be conflict with a previous study using Epstein-Barr virus (EBV)-immortalized FA lymphoid cell lines, which shows that both generation and the overall removal of 8-oxo-G residues in total nuclear DNA is not impaired.Citation40 This discrepancy may be due to completely different methodologies and cell systems used by the 2 studies. First, we measured 8-oxo-dG accumulation specifically in the promoter regions of oxidative stress-sensitive gene CaM1 in primary haematopoietic stem and progenitor cells (LSK cells) by quantitative RT-PCR using DNA after Fpg cleavage, which acts as an N-glycosylase and AP-lysase to excise 8-oxoguanine and other damaged bases, and creates a single-strand break that prevents PCR amplification.Citation11,Citation32 Second, we used the ChIP assay using antibody against 8-oxo-dG. It is in this context that our studies suggest that the FA pathway plays important role in oxidative defense in HSPCs and loss of FA proteins leads to persistent response to oxidative stress.
More recently, we reported a short-lived response of FA HSPCs to oncogenic insults. Our findings indicate that a dysregulated protein arginine methytransferase 5 (PRMT5) in FA HSPCs plays an important role in suppressing p53-dependent tumor surveillance and suggest that PRMT5 may be a putative therapeutic target in FA tumorigenesis.Citation11 In contrast to the short-lived oncogenic response, here we show a persistent oxidative stress-induced response in HSPCs deficient for Fanca or Fancc genes both in vitro and in vivo. Mechanistically, our results indicate that unrepaired DNA damage might be the potential mechanism responsible for long-lasting response to oxidative stress in FA HSPCs. In this context, our studies demonstrate differential responses of HSPCs to oxidative and oncogenic stresses and uncovers an essential role for the FA pathway as an integral part of the versatile cellular mechanism in stress response.
In conclusion, the present study, using an innovative in vivo stress-response model integrating the Gadd45β-luciferase transgene, uncovers 3 novel and significant findings: (i) normal HSPCs can rapidly clear oxidative DNA damage; (ii) FA deficiency renders HSPCs to prolonged oxidative stress response; (iii) the accumulation of unrepaired oxidative DNA damage, particularly in oxidative damage-sensitive genes, may be responsible for the long-lasting response in FA HSPCs. These findings not only provide a molecular explanation for oxidative stress-induced HSC exhaustion in FA, but also suggest novel avenues for therapeutically exploring the pathogenic role of oxidative stress in hematologic diseases.
Materials and methods
Mice and treatment
Fanca+/+, Fanca−/− and Fancc+/+, Fancc−/− mice were generated by interbreeding the heterozygous Fanca+/− or Fancc+/− mice,Citation41,Citation42 respectively. Luc-Fanca+/+ , Luc-Fanca−/− and Luc-Fancc+/+ , Luc-Fancc−/− mice were generated by interbreeding the heterozygous Luc-Gadd45β miceCitation29 with Fanca+/− or Fancc+/− mice.
For paraquat treatment, mice were intraperitoneally (i.p.) injected with increasing doses of paraquat (Sigma-Aldrich, CAS# 75365–73–0, St. Louis, MO) or vehicle. Animals were maintained in the animal barrier facility at Cincinnati Children's Hospital Medical Center (CCHMC) or West Virginia University (WVU). All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of CCHMC and WVU.
Flow cytometry
The lineage marker (Lin) mixture (BD Biosciences, material #: 558074, San Jose, CA) for BM cells from treated or untreated mice included the following biotinylated antibodies: CD3ϵ (145–2C11), CD11b (M1/70), CD45R/B220 (RA3–6B2), mouse erythroid cells Ly-76 (Ter119), Ly6G and Ly-6C (RB6–8C5). Other conjugated antibodies (BD sciences) used for surface staining included: CD45.1 (A20, Cat #: 561871), CD45.2 (A104, Cat#: 560695), Sca1 (D7, Cat#: 561021), c-kit (2B8, Cat#: 560250), CD48 (HM48–1, Cat#: 562398), CD150 (9D1, Cat#: 562647). Biotinylated primary antibodies were detected by incubation of antibody coated cells with streptavidin-PerCP Cy5.5 (Cat#: 551419) or FITC (Cat#: 554060; BD Biosciences) in a 2-step staining procedure.
For apoptosis staining, cells were stained with Annexin V and 7AAD using BD ApoAlert Annexin V kit (BD PharMingen, Cat#: 556547, San Jose, CA) in accordance with the manufacturer's instruction. Apoptosis was analyzed by quantification of Annexin V-positive cell population by flow cytometry.
For cell cycle analysis, surface markers stained cells were stained with propidium iodide (PI, Sigma, Cat#: P4172) containing 1 mg/mL RNase A (Sigma, Cat#: R6513) followed by Flow cytometric analysis.
For γ-H2AX staining, surface marker stained cells were fixed and permeabilized with Cytofix/Cytoperm buffer (BD PharMingen, Cat #: 555028, San Jose, CA) followed by intensive wash using Perm/Wash Buffer (BD PharMingen, Cat #: 555028, San Jose, CA). Cells were then blocked with 2% BSA/PBS for 20 min at room temperature in dark followed by incubation with primary anti-γ-H2AX antibody (biotin-conjugated, Millipore, Cat#: 16–193, Billerica, MA). After washing, cells were then incubated with secondary anti-streptavidin antibody and analyzed by Flow Cytometirc analysis.
Colony-forming unit assay
One,000 LSK cells were plated in a 35-mm tissue culture dish in 4 mL of semisolid medium containing 3 mL of MethoCult M3134 (Stem Cell Technologies, Cat#: 03134, Vancouver, BC, Canada) and the following growth factors: 100 ng/mL SCF (Cat#: 250–03), 10 ng/mL IL-3 (Cat#: 213–13), 100 ng/mL GM-CSF (Cat#: 315–03), and 4 units/mL erythropoietin (Cat#: 100–64) (Peprotech, Burlington, NC). On day 7 after plating, erythroid and myeloid colonies were enumerated. Haematopoietic clonal growth results were expressed as means (of triplicate plates) ± SD of 3 experiments.
Bone marrow transplantation
Paraquat treated or untreated 1,000 LSK cells from WT, Fanca−/− mice along with 3 × 105 BM cells from congenic BoyJ mice were transplanted into lethally irradiated BoyJ recipients.Citation30 4 months post BMT, donor-derived chimera, proportions of LSK cells and SLAM cells in the donor-derived compartment were determined by Flow Cytometric analysis.
To visualize the oxidative stress response in vivo, 1,000 LSK cells from WT mice or 2,000 LSK cells from Luc-Fanca−/− or Luc-Fancc−/− mice along with 3 × 105 BM cells from congenic BoyJ mice were subjected to bone marrow transplantation (BMT) to lethally irradiated BoyJ recipients. 4 months post BMT, the recipients with similar donor-derived chimerism were then subjected to the indicated in vivo treatment and IVIS imaging.
Preparation of cell extracts, immunoblotting and immunoprecipitation
To prepare protein lysates, ∼30,000 sorted LSK cells after treatment were washed with ice-cold PBS, and resuspended in ice-cold lysis buffer containing 50 mmol/L Tris-HCL (pH 7.4), 0.1% NP40, and 1mol/L NaCl supplemented with protease and phosphatase inhibitors [10 μg/mL aprotinin, 25 μg/mL leupeptin, 10 Ag/mL pepstatin A, 2 mmol/L phenylmethylsulfonyl fluoride, 0.1mol/L NaP2O4, 25 mmol/L NaF, and 2 mmol/L sodium orthovandate] for 30 min on ice. Cell debris was removed from the lysates by centrifuging them at 14,000 rpm for 30 min. Protein concentration was quantified by using Bio-Rad reagent and resolved on SDS-PAGE and transferred onto nitrocellulose membranes. Immunoblots were then incubated with primary antibodies specific for γ-H2AX (Upstate Biotechnology, Cat#: 05–636, Danvers, MA) and β-actin (Sigma-Aldrich, Cat#: A5441), respectively.
IVIS imaging
Subjected mice pre-treated with indicated chemicals were injected by an intraperitoneal route with a luciferin solution (15 mg/ml in PBS (150 mg/kg)) that is allowed to distribute in awake animals for about 5–15 minutes.Citation11,Citation30 The mice are placed into a clear plexiglass anesthesia box (2.5–3% isofluorane) that allows unimpeded visual monitoring of animals. After the mice are fully anesthetized, they are transferred from the box to the nose cones attached to the manifold in the imaging chamber. The first image is taken approximately 5 minutes after luciferin injection. Continue to take images at indicated time points after the injection using IVIS series pre-clinical in vivo Imaging systems (PerkinElmer, Santa Clara, CA). The bioluminescence image signals were quantified by measuring photon flux using LiveImage Pro. 2.0 software (Caliper Life Science, Hopkinton, MA).
Comet assay
The generation of DNA strand breaks was assessed by the single cell gel electrophoresis (Comet) assay, using a Fpg-FLARE (fragment length analysis using repair enzymes) comet assay kit in accordance with the manufacturer's instructions (Trevigen, Cat#: 4250–050-K, Gaithersburg, MD). For each experimental point at least 3 different cultures were analyzed, and 300 cells were evaluated from each culture. Comet tail length and tail moment were measured under a fluorescence microscope (Zeiss Model C4742–80–12AG) using an automated image analysis system based on a public domain NIH Image program.
Eight-oxodG incorporation assay
Eight-oxodG incorporation assay was performed as described previously.Citation32 Briefly, 250 ng of genomic DNA was incubated with 8-oxoguaine-specific N-glycosylase human OGG1 (New England Biolabs, Cat #: M0241S, Ipswich, MA) in 1X NEBuffer 1 (10 mM Bis Tris Propane-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.0) and 100 µg/ml BSA in a volume of 50 μl at 37°C for 12hr. OGG1 enzyme was then inactivated by incubation at 60°C for 5 min. The reaction mixture was then used for a quantitative PCR assay using primers for calmodulin 1 (CaM1) and GAPDH.
Chromatin immunoprecipitation
The ChIP assay was performed as described previouslyCitation32 with minor modifications. Briefly, chromatin was cross-linked by adding 37% formaldehyde with rotation at 4°C for 10 minutes and then room temperature for 20 minutes. The pellet was resuspended in lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-HCL, pH 8.1, and protease inhibitors) and sonicated with repeated 10-second pulses. Residual unfragmented chromatin was removed by centrifugation at 15 000 g for 10 minutes. The amount of DNA in the supernatant was quantified and adjusted to 100 ng/μL. Supernatant (200 μL) was diluted 10-fold in 2 mL of ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2mM EDTA, 16.7mM Tris-HCL, pH 8.1, 167mM NaCl, and protease inhibitors), and precleaned twice with BSA-blocked protein L agarose (Pierce, Cat#: 20520, Rockford, IL). The beads were centrifuged and the supernatant was divided into 500-μL aliquots representing input DNA, and material for immunoprecipitation, and an IgG control. Primary antibody was added and incubated at 4°C overnight. Anti-8-oxoguanine (Chemicon, Cat#: MAB3560, Billerica MA), antibody was used for immunoprecipitation, and ChromPure rabbit IgG (Jackson ImmunoResearch Laboratories, Cat#: 011–000–003, West Grove, PA) was used for the IgG control. Precipitated samples were then subjected to PCR using primers specific for promoter regions of Calmodulin 1 (CaM1) gene.
Molecular cloning and lentivirus production
For lentiviral vector construction, full-length FANCA cDNA was cloned into pRRL-SIN-cPPT-MNDU3-MCS-IVW (TMND-IRES-Venus) vector.Citation30,Citation33 The plasmids (10 μg each) were used to produce retroviral supernatant.
Statistics
Data were analyzed statistically using a Student's t test. The level of the statistical significance stated in the text was based on the P values. P < 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1320627_Supplemental_Material.docx
Download MS Word (569.4 KB)Acknowledgments
We thank Dr. Madeleine Carreau (Laval University) for Fanca+/− mice, Dr. Manuel Buchwald (University of Toronto) for Fancc+/− mice, Dr. Punam Malik (Cincinnati Children's Hospital Medical Center, CCHMC) for TMND-IRES-Venus vector.
Funding
This work was supported by the National Natural Science Foundation of China (NNSFC #81470288; 81370608 and U1401221). W.D. is supported by West Virginia University (WVU) Health Science Center (HSC) and School of Pharmacy (SOP) startup package, and West Virginia Clinical and Translational Science Institute (WV CTSI) funding (C1006000DW).
Related Research Data
References
- Bagby GC Jr. Genetic basis of Fanconi anemia. Curr Opin Hematol 2003;10(1):68-76; PMID:12483114; https://doi.org/10.1097/00062752-200301000-00011
- Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d'Enghien C, Bluteau O, Cuccuini W, Gachet S, Peffault de Latour R, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 2016;126:3580-4; PMID:27500492; https://doi.org/10.1172/JCI88010
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011;11(7):467-80; PMID:21701511; https://doi.org/10.1038/nrc3088
- Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics 2015; 9:32; PMID:26596371; https://doi.org/10.1186/s40246-015-0054-y
- Du W, Erden O Pang Q. TNF-a signaling in Fanconi anemia. Blood Cells Mol Dis 2013; 52(1):2-11; PMID:23890415; https://doi.org/10.1016/j.bcmd.2013.06.005
- Kennedy RD, D'Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 2005; 19(24):2925-40; PMID:16357213; https://doi.org/10.1101/gad.1370505
- Meyer S, Neitzel H, T¨onnies H. Chromosomal aberrations associated with clonal evolution and leukemic transformation in Fanconi anemia: clinical and biological implications. Anemia 2012: 2012:349837; PMID:22675616; https://doi.org/10.1155/2012/349837
- Park JY, Virts EL, Jankowska A, Wiek C, Othman M, Chakraborty SC, Vance GH, Alkuraya FS, Hanenberg H, Andreassen PR. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet 2016; 53:672-80; PMID:27208205; https://doi.org/10.1136/jmedgenet-2016-103847
- Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium, Majewski J, Dyment DA, Innes AM, et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov 2015; 5(2):135-42; PMID:25472942; https://doi.org/10.1158/2159-8290.CD-14-1156
- Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet 2003; 40(1):1-10; PMID:12525534; https://doi.org/10.1136/jmg.40.1.1
- Du W, Amarachintha S, Erden O, Wilson A, Pang Q. The Fanconi anemia pathway controls oncogenic response in hematopoietic stem and progenitor cells by regulating PRMT5-mediated p53 arginine methylation. Oncotarget 2016; 7(37):60005-20; PMID:27507053; https://doi.org/10.18632/oncotarget.11088
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493:356-63; PMID:23325218; https://doi.org/10.1038/nature11863
- Freidovich I. Fundamental aspects of reactive oxygen species, or what's the matter with oxygen? NY Acad Sci 1999; 893:13; PMID:10672226; https://doi.org/10.1111/j.1749-6632.1999.tb07814.x
- Fang Y, Sheng Y, Guoyao Wu. Free radicals, antioxidants, and nutrition. Nutrition 2002; 18:872-9; PMID:12361782; https://doi.org/10.1016/S0899-9007(02)00916-4
- Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal 2008; 10(11):1909-21; PMID:18627348; https://doi.org/10.1089/ars.2008.2129
- Buchwald M, Moustacchi E. Is Fanconi anemia caused by a defect in the processing of DNA damage? Mutat Res 1998; 408 (2):75-90; PMID:9739810
- Fagerlie SR, Diaz J, Christianson TA, McCartan K, Keeble W, Faulkner GR, Bagby GC. Functional correction of FA-C cells with FANCC suppresses the expression of interferon gamma-inducible genes. Blood 2001; 97(10):3017-24; PMID:11342426; https://doi.org/10.1182/blood.V97.10.3017
- Du W, Rani R, Sipple J, Schick J, Myers KC, Mehta P, Andreassen PR, Davies SM, Pang Q. The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters. Blood 2012; 119(18):4142-51; PMID:22408259; https://doi.org/10.1182/blood-2011-09-381970
- Pang Q, Andreassen PR. Fanconi anemia proteins and endogenous stresses. Mutat Res 2009; 668(1–2):42-53; PMID:19774700; https://doi.org/10.1016/j.mrfmmm.2009.03.013
- Li J, Sejas DP, Zhang X, Qiu Y, Nattamai KJ, Rani R, Rathbun, KR, Geiger H, Williams DA, Bagby GC, et al. TNF-a induces leukemic clonal evolution ex vivo in Fanconi anemia group C stem cells. J Clin Invest 2007; 117:3283-95; PMID:17960249; https://doi.org/10.1172/JCI31772
- Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol 1997; 15:351-69; PMID:9143692; https://doi.org/10.1146/annurev.immunol.15.1.351
- Sejas DP, Rani R, Qiu Y, Zhang X, Fagerlie SR, Nakano H, Williams DA, Pang Q. Inflammatory reactive oxygen species-mediated hematopoietic suppression in Fancc-deficient mice. J Immunol 2007; 178:5277-87; PMID:17404312; https://doi.org/10.4049/jimmunol.178.8.5277
- Zhang X, Sejas DP, Qiu Y, Williams DA, Pang Q. Inflammatory ROS promote and cooperate with Fanconi anemia mutation for hematopoietic senescence. J Cell Science 2007; 120:1572-83; PMID:17405815; https://doi.org/10.1242/jcs.003152
- Cumming RC, Liu JM, Youssoufian H, Buchwald M. Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood 1996; 88 (12):4558-67; PMID:8977247
- Otsuki T, Nagakura S, Wang J, Bloom M, Grompe M, Liu JM. Tumor necrosis factor-alpha and CD95 ligation suppress erythropoiesis in Fanconi anemia C gene knockout mice. J Cell Physiol 1999; 179 (1):79-86; PMID:10082135; https://doi.org/10.1002/(SICI)1097-4652(199904)179:1<79::AID-JCP10>3.0.CO;2-O
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013; 339(6116):211-4; PMID:23223453; https://doi.org/10.1126/science.1227166
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005; 121:1109-21; PMID:15989959; https://doi.org/10.1016/j.cell.2005.05.026
- Bogliolo M, Cabré O, Callén E, Castillo V, Creus A, Marcos R, and Surrallés J. The Fanconi anaemia genome stability and tumour suppressor network. Mutagenesis 2002; 17(6):529-38; PMID:12435850; https://doi.org/10.1093/mutage/17.6.529
- Zhang N, Ahsan MH, Zhu L, Sambucetti LC, Purchio AF, West DB. NF-kappaB and not the MAPK signaling pathway regulates GADD45beta expression during acute inflammation. Biol Chem 2005; 280(22):21400-08; https://doi.org/10.1074/jbc.M411952200
- Du W, Amarachintha S, Wilson A, Pang Q. The immune receptor Trem1 cooperates with diminished DNA damage response to induce preleukemic stem cell expansion. Leukemia 2017; 31(2):423-33; PMID:27568523; https://doi.org/10.1038/leu.2016.242
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003; 114:371-83; PMID:12914701; https://doi.org/10.1016/S0092-8674(03)00567-1
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature 2004;429(6994):883-91; PMID:15190254; https://doi.org/10.1038/nature02661
- Li X, Erden O, Li L, Ye Q, Wilson A, Du W. Binding to WGR domain by salidroside activates PARP1 and protects hematopoietic stem cells from oxidative stress. Antioxid Redox Signal 2014; 20(12):1853-65; PMID:24294904; https://doi.org/10.1089/ars.2013.5600
- Saadatzadeh MR, Bijangi-Vishehsaraei K, Hong P, Bergmann H, Haneline LS. Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. J Biol Chem 2004; 279:16805-12; PMID:14764578; https://doi.org/10.1074/jbc.M313721200
- Hadjur S, Ung K, Wadsworth L, Dimmick J, Rajcan-Separovic E, Scott RW, Buchwald M, Jirik FR. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood 2001; 98:1003-11; PMID:11493445; https://doi.org/10.1182/blood.V98.4.1003
- Cumming RC, Lightfoot J, Beard K, Youssoufian H, O'Brien PJ, Buchwald M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med 2001; 7:814-20; PMID:11433346; https://doi.org/10.1038/89937
- Kruyt FA, Hoshino T, Liu JM, Joseph P, Jaiswal AK, Youssoufian H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood 1998; 2:3050-56; PMID:9787138
- Mukhopadhyay SS, Leung KS, Hicks MJ, Hastings PJ, Youssoufian H, Plon SE. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J Cell Biol 2006; 175:225-35; PMID:17060495; https://doi.org/10.1083/jcb.200607061
- Park SJ, Ciccone SL, Beck BD, Hwang B, Freie B, Clapp DW, Lee SH. Oxidative stress/damage induces multimerization and interaction of Fanconi anemia proteins. J Biol Chem 2004; 279:30053-59; PMID:15138265; https://doi.org/10.1074/jbc.M403527200
- Will O, Schindler D, Boiteux S, Epe B. Fanconi's anaemia cells have normal steady-state levels and repair of oxidative DNA base modifications sensitive to Fpg protein. Mutat Res 1998; 409(2):65-72; PMID:9838922; https://doi.org/10.1016/S0921-8777(98)00043-3
- Chen M, Tomkins DJ, Auerbach W, McKerlie C, Youssoufian H, Liu L, Gan O, Carreau M, Auerbach A, Groves T, et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. See comment in PubMed Commons belowNat Genet 1996; 12(4):448-51; PMID:8630504; https://doi.org/10.1038/ng0496-448
- Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 2003; 12:2063-76; PMID:12913077; https://doi.org/10.1093/hmg/ddg219