
ABSTRACT
For almost a decade, there has been much interest in the development of chemical inhibitors of Polo-like kinase 1 (Plk1) protein interactions. Plk1 is a master regulator of the cell division cycle that controls numerous substrates. It is a promising target for cancer drug development. Inhibitors of the kinase domain of Plk1 had some success in clinical trials. However, they are not perfectly selective. In principle, Plk1 can also be inhibited by interfering with its protein interaction domain, the Polo-Box Domain (PBD). Selective chemical inhibitors of the PBD would constitute tools to probe for PBD-dependent functions of Plk1 and could be advantageous in cancer therapy. The discovery of Poloxin and thymoquinone as PBD inhibitors indicated that small, cell-permeable chemical inhibitors could be identified. Other efforts followed, including ours, reporting additional molecules capable of blocking the PBD. It is now clear that, unfortunately, most of these compounds are non-specific protein alkylators (defined here as groups covalently added via a carbon) that have little or no potential for the development of real Plk1 PBD-specific drugs. This situation should be minded by biologists potentially interested in using these compounds to study Plk1. Further efforts are needed to develop selective, cell-permeable PBD inhibitors.
The PBD as a drug target
Plk1 is a master regulator of cell division.Citation1,Citation2 In addition to a Ser/Thr kinase domain, it contains a C-terminal protein interaction domain, the PBD.Citation3 The PBD allows Plk1 to bind several proteins, often via a phosphorylated motif (S-pS/pT) that docks into its phospho-binding pocket.Citation4 In many cases, this binding allows Plk1 to phosphorylate the bound protein at another site to modify its activity (). It also allows Plk1 to dock to protein complexes and structures that it regulates during cell division, including centrosomes, kinetochores and the cytokinetic midbody.Citation1,Citation2 In addition, the PBD can interact with and inhibit the kinase domain (KD) of Plk1 intramolecularly.Citation5,Citation6 Although recognition by the PBD may not be required for the phosphorylation of all Plk1 substrates, the function of the PBD is essential for cell division.Citation7,Citation8
Figure 1. Schematic overview of Plk1 function. A. The Polo-Box Domain (PBD) mediates protein interactions with targets (X). These interactions are often enhanced by priming phosphorylation (yellow). This docking facilitates the phosphorylation (orange) of the same target or another target in the vicinity (Y) by the kinase domain (KD). B. A competitive chemical inhibitor (red) of the PBD prevents Plk1 from interacting with its targets, thereby decreasing the efficiency of substrate phosphorylation by the KD.
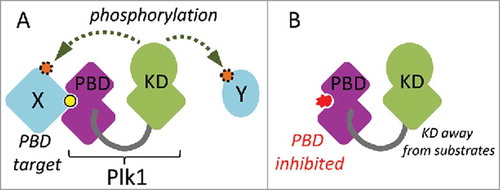
Plk1 is considered as a good cancer drug target because it is essential for cell division and because many types of cancer cells rely on a higher level of Plk1 activity to divide and survive than non-transformed cells.Citation9-Citation13 Inhibitors of the KD of Plk1 have yielded promising results in clinical trials, but the doses that can be given to patients are limited by the toxicity of the compounds.Citation13 As these inhibitors are not completely selective for Plk1, their toxicity could be partly due to their interference with other kinases. For instance, volasertib (BI 6727), an inhibitor of Plk1 that gave promising results for some types of Acute Myeloid Leukemia, also inhibits Plk2 and Plk3 with similar IC50 values.Citation14,Citation15 This may be problematic in the context of cancer treatment as Plk2 and Plk3 function in mechanisms that prevent cell cycle progression in the presence of DNA damage.Citation1 In principle, the PBD constitutes a second drug target within Plk1. PBDs exists only in the 5 members of the Polo-Like Kinase (PLK) family.Citation1,Citation16 Moreover, the PBD differs more between the PLKs than the KD does, and PBD binding motifs differ between PLKs.Citation17 Therefore, chemical inhibition of the PBD of Plk1 is considered a promising avenue for the development of more selective cancer drugs targeting Plk1.Citation12
Efforts in the development of drug-like PBD inhibitors – several alkylators found
In 2008, Reindl et al published the discovery of the first small-molecule inhibitors of the PBD of Plk1.Citation18 In an in vitro chemical screen using a fluorescence polarization (FP) assay, they identified Poloxin () as a chemical capable of interfering with the interaction between the PBD and an optimal phosphopeptide. They subsequently found that thymoquinone (TQ), a chemically related and natural molecule, had the same effect, with a similar potency in the low micromolar range. Higher concentrations of either compound were required to inhibit cell proliferation and cell toxicity was problematic.Citation18
Figure 2. Structures of published PBD/Plk1 inhibitors. Only the inhibitors discussed in the text are shown. Arrows indicate sites of nucleophilic attacks by amino-acid side chains leading to a covalent bond (alkylation of the protein). Shown is the potency (IC50) of each molecule for the inhibition of PBD domains measured in fluorescence polarization assays or GST pulldown (Purpurogallin). See indicated references for details. Rigosertib is reported as a non-ATP-competitive inhibitor of Plk1 kinase and has not been shown to interfere with the PBD. The asterisk indicates a suspected site of nucleophilic attack.
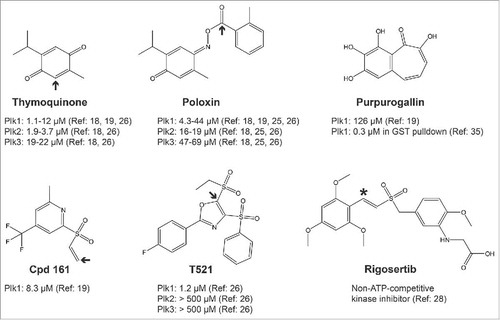
We decided to develop a cell-based assay allowing the identification of PBD inhibitors with the hope that it would facilitate the immediate detection of membrane-permeable compounds active in the cell. The assay uses Bioluminescence Resonance Energy Transfer (BRET), in which Plk1 is fused to Luciferase and a PBD-interacting protein is fused to GFP.Citation19,Citation20 When both proteins interact, energy is transferred from Luciferase to GFP, which fluoresces. Compounds identified as BRET inhibitors were then tested for their ability to interfere with mitosis as expected for Plk1 PBD inhibitors. Only 2 chemotypes were effective in this test. Subsequent biochemical assays including the FP assay of Reindl et al (2008), which monitors the interaction of the PBD with an optimal phosphopeptide, validated only one compound as an effective inhibitor of PBD function at low micromolar concentrations.Citation19,Citation21,Citation22 However, Structure-Activity Relationship (SAR) studies on this molecule revealed that it spontaneously cleaves to generate a vinyl sulfone function that is a powerful alkylator of any nucleophilic amino-acid side chain (Cpd 161, , here alkylator is defined as any group covalently added via a carbon). We showed that it reacts with amino-protected lysines, histidines and cysteines and we detected multiple alkylation sites in the PBD of Plk1 after in vitro reaction. We used liquid chromatography-tandem mass spectrometry (LC-MS/MS) to map alkylation sites on the PBD. Although some of the detected sites were in or near the canonical PBD binding site, other alkylated residues were located far from it, all over the protein.Citation19
Because TQ and Poloxin behaved similarly to Cpd 161 in our cell-based and in vitro assays, we wondered if, like Cpd 161, they were alkylators. This possibility was suggested already in the initial report by Reindl et al, based on the fact that PBD inhibition by TQ and Poloxin was time-dependent.Citation18 It was also supported by the chemical structures of these compounds which seemed susceptible to nucleophilic attacks.Citation23 Moreover, a modeling study had proposed that TQ and Poloxin could potentially form a covalent bond within the phospho-binding pocket of the PBD.Citation24 Finally, the idea that Poloxin reacted by benzoylation with the PBD had been further reinforced by the observations that the activated ester group of Poloxin and analogs is essential for PBD inhibitory activity.Citation25 A fluorescent Poloxin derivative also allowed Scharow et al to determine that, to bind the PBD, Poloxin does not require the PBD amino-acid residues known to be crucial for its phospho-binding pocket.Citation25 Attempts by these authors and by us to map binding or alkylation sites on the PBD using NMR failed for technical reasons.
Using LC-MS/MS, we identified alkylation sites by TQ and Poloxin (in parallel with Cpd 161) on the PBD.Citation19 While alkylated cysteine and lysine residues were found after reaction with TQ, only lysine residues were mapped with Poloxin. This specificity is consistent with the reactions we observed with individual amino-protected amino-acids. As for Cpd 161, alkylated sites detected were distant from the PBD phospho-binding site.
More recently, Chen et al reported the identification of T521, another compound capable of inhibiting the PBD of Plk1 by alkylation, again outside the phospho-binding pocket.Citation26 Its structure is different from that of Cpd 161, but nevertheless, contains an activated carbon double bond adjacent to a sulfone group, like Cpd 161 (). In fact, the double-bonded carbons are flanked by 2 sulfone groups in T521. One is an ethyl sulfone that is considered a good leaving group that can be substituted by the amino group of a lysine residue.Citation26
Although Poloxin, TQ, Cpd 161 and T521 do cause cellular arrests or delays in mitosis, apoptosis and decreased proliferation,Citation18,Citation19,Citation26 which are the results expected upon Plk1 inhibition, it is doubtful that these effects result solely from Plk1 inhibition. These molecules probably alkylate multiple cellular proteins on exposed nucleophilic side-chains. Disruption of the activities of many of them, such as tubulins or mitotic enzymes, would cause mitotic problems and arrests. The same can be said about anti-tumor effects observed in mouse models.Citation26,Citation27 For these reasons, it is unreasonable to use these unspecific alkylators to probe the cellular effects of Plk1 PBD inhibition. Likewise, these molecules do not constitute promising starting points for targeted cancer drugs.
Interestingly, ON01910, a non-ATP-competitive inhibitor of Plk1 also known as Rigosertib,Citation28 contains a carbon double bond next to a sulfone group, like the alkylating functions in Cpd 161 and T521 (). We did not detect reaction of Rigosertib with amino-protected cysteine in vitro (J.-F. Lavallée, personal communications), but its reactivity has not been examined in depth. Rigosertib was already found to inhibit several unrelated kinases and to interfere with microtubules polymerization.Citation28,Citation29
Can an alkylator be a selective PBD inhibitor?
The conclusion that TQ inhibits Plk1 by non-specific alkylation of nucleophilic amino-acid residues may be surprising since it was shown by crystallography that it binds the PBD as a phospho-mimic at its canonical phospho-binding site.Citation30 However, it should be kept in mind that this co-crystal structure was obtained after soaking PBD crystals in a TQ-containing solution. In this context, TQ may have a relatively high affinity for the phospho-binding pocket, compared with other sites in the packed protein crystals. When binding of TQ or Poloxin to the PBD is modeled using probabilistic calculations, they are predicted to bind preferentially in the phospho-binding pocket, of all places.Citation24 Similarly, Cpd 161, which clearly inhibits the PBD by alkylation via its vinyl group, was predicted by unbiased modeling to bind the same phospho-binding pocket non-covalently and independently from the vinyl group.Citation19 To us, these results highlight the limitations of these approaches. In both cases, the small molecule is forced to find a binding site on the protein, and it is to be expected that the stickiest spot naturally tends to be the protein interaction site. However, in the cell, this higher affinity may not be significant.
For an alkylator to bear potential as a target-specific inhibitor, it seems reasonable to expect its scaffold to have significant specific affinity for the target already without the alkylating chemical function. There have been cases where the potency of pre-existing inhibitors of a target protein was increased by the addition of an alkylating group that renders the inhibitor irreversible (for example, see ref31). However, this has not been documented for a PBD inhibitor. In the case of Cpd 161, analogs with a saturated vinyl were inactive in vitro at the highest concentrations tested of 300 μM.Citation19
Despite the fact that several compounds identified so far are alkylators, some of them have been shown to inhibit the PBD of Plk1 with lower IC50 values compared with other protein interaction domains (). For example, Poloxin inhibits the PBDs of Plk1 and Plk3 with IC50 values around 5 μM and 50 μM, respectively.Citation18,Citation26 In the case of T521, the selectivity is even more pronounced, with IC50 values around 1 μM for the PBD of Plk1 and above 500 μM, for the PBDs of Plk2 and Plk3.Citation26 These differences could be due to the presence of more reactive nucleophilic side chains (because of their immediate environments) in the PBD of Plk1 relative to the PBD of other PLKs or other protein interaction domains. It could also be due to the potential location of alkylation sites in the PBD of Plk1 at sites that cause steric hindrance or a minor conformational change that results in inhibition, a phenomenon that would not necessarily occur in other alkylated proteins. Finally, it is possible that the PBD of Plk1 is grossly unfolded following alkylation, and that this does not happen with the other proteins tested. In favor of the latter possibility, alkylation of the PBD of Plk1 by T521 was found to cause a major change in conformation as detected by circular dichroism.Citation26 Incidentally, we found the PBD of Plk1 to be extremely sensitive to inactivation by heat and by freeze-thaw cycles (our unpublished observations). Whether the PBD of Plk2 or Plk3 are resistant to alkylation-induced major conformation shift or unfolding was not tested.
Even if a protein alkylator preferentially interferes with Plk1 vs Plk2 or Plk3, the inhibition of countless untested proteins could potentially contribute to the cellular phenotypes observed. The only way to prove that a compound induces a cellular phenotype through its action on a particular target is to rescue this phenotype by expressing a mutant version of the protein that is resistant to the compound but retains its normal cellular function. In principle, this type of experiment is much more difficult for competitive inhibitors than for non-competitive inhibitors.
Outlook
As for most protein interactions, a major difficulty in developing a chemical inhibitor of the PBD is that it must compete with another protein for binding on a large and topologically complex surface on the PBD. The most potent and selective inhibitors of the PBD obtained so far have been peptide-like molecules.Citation12,Citation32,Citation33 However, they tend to be poorly permeable to the plasma membrane due to their large size and the presence of charged groups such as a phosphate.Citation32,Citation34 It may be possible to circumvent this problem by using innovative strategies to enhance the delivery of these compounds to the cell, or by replacing charged or polar groups in the peptidoid with less lipophobic groups.Citation12
The development of bioavailable small-molecule inhibitors of PBD interactions has been very challenging. No such compound documented yet is potent and selective enough to constitute a potential cancer drug or even be useful as a tool in cell biology. Although we have focused our discussion on alkylating agents, not all published PBD-interfering compounds fall in this category, but they all lack in potency or selectivity. For example, the natural benzotropolone compound purpurogallin (PPG) has been identified as a low-micromolar Plk1 PBD inhibitor in an in vitro screen, and to cause mitotic blocks and apoptosis in cell culture.Citation35 However, although it is unlikely to modify proteins covalently, PPG interferes with various other proteins. A variety of structural features, including poly-hydroxylation as in PPG, have been shown to render compounds particularly likely to be recovered non-specifically in high-throughput screening assays. Such compounds have been termed Pan-Assay INterfering compounds (PAINs).Citation36 Perhaps drug screens using much larger chemical libraries or using a fragment-based approach, combined with structure-assisted virtual screening and rational design will help develop more potent, selective and bioavailable PBD inhibitors.Citation37
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgement
We thank Jean-François Lavallée of IRIC's medicinal chemistry platform for useful discussions and comments on the manuscript.
Funding
Work on the development of Plk1 inhibitors in the Archambault lab was funded by a grant from the Canadian Cancer Society Research Institute. VA holds a Chercheur boursier Junior 2 salary award from the Fonds de recherche du Québec – Santé (FRQS). IRIC is supported in part by the Canada Foundation for Innovation and by the FRQS.
Related Research Data
References
- Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 2014; 15:433-52; PMID:24954208; https://doi.org/10.1038/nrm3819
- Archambault V, Lepine G, Kachaner D. Understanding the Polo Kinase machine. Oncogene 2015; 34:4799-807; PMID:25619835; https://doi.org/10.1038/onc.2014.451
- Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003; 299:1228-31; PMID:12595692; https://doi.org/10.1126/science.1079079
- Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003; 115:83-95; PMID:14532005; https://doi.org/10.1016/S0092-8674(03)00725-6
- Jang YJ, Lin CY, Ma S, Erikson RL. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc Natl Acad Sci U S A 2002; 99:1984-9; PMID:11854496; https://doi.org/10.1073/pnas.042689299
- Xu J, Shen C, Wang T, Quan J. Structural basis for the inhibition of Polo-like kinase 1. Nat Struct Mol Biol 2013; 20:1047-53; PMID:23893132; https://doi.org/10.1038/nsmb.2623
- Hanisch A, Wehner A, Nigg EA, Sillje HH. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol Biol Cell 2006; 17:448-59; PMID:16267267; https://doi.org/10.1091/mbc.E05-08-0801
- Lera RF, Burkard ME. High mitotic activity of Polo-like kinase 1 is required for chromosome segregation and genomic integrity in human epithelial cells. J Biol Chem 2012; 287:42812-25; PMID:23105120; https://doi.org/10.1074/jbc.M112.412544
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137:835-48; PMID:19490893; https://doi.org/10.1016/j.cell.2009.05.006
- Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26:2093-108; PMID:16507989; https://doi.org/10.1128/MCB.26.6.2093-2108.2006
- Renner AG, Dos Santos C, Recher C, Bailly C, Creancier L, Kruczynski A, Payrastre B, Manenti S. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009; 114:659-62; PMID:19458358; https://doi.org/10.1182/blood-2008-12-195867
- Lee KS, Burke TR Jr, Park JE, Bang JK, Lee E. Recent advances and new strategies in targeting Plk1 for anticancer therapy. Trends Pharmacol Sci 2015; 36:858-77; PMID:26478211; https://doi.org/10.1016/j.tips.2015.08.013
- Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther 2016; 15:1427-35; PMID:27330107; https://doi.org/10.1158/1535-7163.MCT-15-0897
- Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015; 29:11-9; PMID:25027517; https://doi.org/10.1038/leu.2014.222
- Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res 2009; 15:3094-102; PMID:19383823; https://doi.org/10.1158/1078-0432.CCR-08-2445
- de Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle 2011; 10:2255-62; PMID:21654194; https://doi.org/10.4161/cc.10.14.16494
- van de Weerdt BC, Littler DR, Klompmaker R, Huseinovic A, Fish A, Perrakis A, Medema RH. Polo-box domains confer target specificity to the Polo-like kinase family. Biochim Biophys Acta 2008; 1783:1015-22; PMID:18359294; https://doi.org/10.1016/j.bbamcr.2008.02.019
- Reindl W, Yuan J, Kramer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008; 15:459-66; PMID:18482698; https://doi.org/10.1016/j.chembiol.2008.03.013
- Normandin K, Lavallee JF, Futter M, Beautrait A, Duchaine J, Guiral S, Marinier A, Archambault V. Identification of Polo-like kinase 1 interaction inhibitors using a novel cell-based assay. Sci Rep 2016; 5:37581; PMID:27874094; https://doi.org/10.1038/srep37581
- Bacart J, Corbel C, Jockers R, Bach S, Couturier C. The BRET technology and its application to screening assays. Biotechnol J 2008; 3:311-24; PMID:18228541; https://doi.org/10.1002/biot.200700222
- Reindl W, Strebhardt K, Berg T. A high-throughput assay based on fluorescence polarization for inhibitors of the polo-box domain of polo-like kinase 1. Anal Biochem 2008; 383:205-9; PMID:18793607; https://doi.org/10.1016/j.ab.2008.08.014
- Reindl W, Strebhardt K, Berg T. A fluorescence polarization assay for the discovery of inhibitors of the polo-box domain of polo-like kinase 1. Methods Mol Biol 2012; 795:69-81; PMID:21960216
- Zhou Y, Jianhua C, Rehse PH. Thymoquinone and Poloxin are slow-irreversible inhibitors to human Polo-like kinase 1 Polo-box domain. J Med Colleges PLA 2010; 25:136-42; https://doi.org/10.1016/S1000-1948(10)60032-9
- Liao C, Park JE, Bang JK, Nicklaus MC, Lee KS. Exploring potential binding modes of small drug-like molecules to the polo-box domain of human polo-like kinase 1. ACS Med Chem Lett 2010; 1:110-4; PMID:20625469; https://doi.org/10.1021/ml100020e
- Scharow A, Raab M, Saxena K, Sreeramulu S, Kudlinzki D, Gande S, Dötsch C, Kurunci-Csacsko E, Klaeger S, Kuster B, et al. Optimized Plk1 PBD Inhibitors Based on Poloxin Induce Mitotic Arrest and Apoptosis in Tumor Cells. ACS Chem Biol 2015; 10:2570-9; PMID:26279064; https://doi.org/10.1021/acschembio.5b00565
- Chen Y, Zhang J, Li D, Jiang J, Wang Y, Si S. Identification of a novel Polo-like kinase 1 inhibitor that specifically blocks the functions of Polo-Box domain. Oncotarget 2016; 8(1):1234-46.
- Yuan J, Sanhaji M, Kramer A, Reindl W, Hofmann M, Kreis NN, Zimmer B, Berg T, Strebhardt K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol 2011; 179:2091-9; PMID:21839059; https://doi.org/10.1016/j.ajpath.2011.06.031
- Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005; 7:275-86; PMID:15766665; https://doi.org/10.1016/j.ccr.2005.02.009
- Lu T, Laughton CA, Wang S, Bradshaw TD. In vitro antitumor mechanism of (E)-N-(2-methoxy-5-(((2,4,6-trimethoxystyryl)sulfonyl)methyl)pyridin-3-yl)methane sulfonamide. Mol Pharmacol 2014; 87:18-30; PMID:25316768; https://doi.org/10.1124/mol.114.093245
- Yin Z, Song Y, Rehse PH. Thymoquinone blocks pSer/pThr recognition by Plk1 Polo-box domain as a phosphate mimic. ACS Chem Biol 2013; 8:303-8; PMID:23135290; https://doi.org/10.1021/cb3004379
- Anscombe E, Meschini E, Mora-Vidal R, Martin MP, Staunton D, Geitmann M, Danielson UH, Stanley WA, Wang LZ, Reuillon T, et al. Identification and characterization of an irreversible inhibitor of CDK2. Chem Biol 2015; 22:1159-64; PMID:26320860; https://doi.org/10.1016/j.chembiol.2015.07.018
- Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat Chem Biol 2011; 7:595-601; PMID:21765407; https://doi.org/10.1038/nchembio.614
- Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR Jr. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem Biol 2012; 7:805-10; PMID:22292814; https://doi.org/10.1021/cb200469a
- Qian WJ, Park JE, Lim D, Lai CC, Kelley JA, Park SY, Lee KW, Yaffe MB, Lee KS, Burke TR Jr. Mono-anionic phosphopeptides produced by unexpected histidine alkylation exhibit high Plk1 polo-box domain-binding affinities and enhanced antiproliferative effects in HeLa cells. Biopolymers 2014; 102:444-55; PMID:25283071; https://doi.org/10.1002/bip.22569
- Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem 2009; 284:2344-53; PMID:19033445; https://doi.org/10.1074/jbc.M805308200
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2012; 53:2719-40; https://doi.org/10.1021/jm901137j
- Sheng C, Dong G, Miao Z, Zhang W, Wang W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem Soc Rev 2015; 44:8238-59; PMID:26248294; https://doi.org/10.1039/C5CS00252D