
ABSTRACT
Much effort is currently focused on the p53 pathway. p53 is a key tumor suppressor, which is mutated or lost in many human cancers. Restoration of the p53 pathway holds the potential to induce selective cell death in tumor cells without harming normal cells that have intact p53 pathways. Most tumor cells express mutated p53 or suppress p53 by overexpression of MDM2. In this study, a compound referred to as CB002 with one closely related compound from the Chembridge library were evaluated for tumor cytotoxicity without affecting normal cells by restoration of the p53 pathway. A decrease of mutant p53 protein expression, restoration of inactivated p53, or some activation of p73 are candidate mechanisms this agent could cause tumor cell apoptosis and growth arrest. We further show that CB002 activates p53 pathway signaling in part via p73 in p53 mutant cancer cell lines. However, it is important to note that we have not established a role for p73 in the anti-tumor effect of CB002 or R1. CB002 causes tumor cell death with synergistic effects with traditional chemotherapeutics CPT-11 and 5-FU.
Introduction
The p53 pathway is one of the most critical pathways in antitumor drug development. p53 is mutated with missense mutations in over half of all human tumors.Citation1,Citation2 Wild-type p53 was shown conclusively in 1989 to be a potent tumor suppressor after initial research suggested it was a proto-oncogene.Citation3 The confusing early research on p53 has been shown to be due to mutant p53 exhibiting opposing effects on cell proliferation compared with wild-type.Citation3 Mutant p53 proteins exhibit a wide variety of changes in response depending on the mutation, meaning that all p53 mutations are not equivalent.Citation3 Wild-type p53 is typically constitutively expressed at low levels with rapid turnover due to mouse double minute 2 (MDM2) E3 ubiquitin ligase, while mutant p53 is often expressed at extremely high levels in tumor cells.Citation2,Citation3
MDM2 overexpression is another common route through which the p53 pathway is suppressed in tumor cells.Citation1 MDM2 normally forms a negative feedback loop with p53 by transcriptional activation of MDM2 via p53, and MDM2 causing ubiquitin mediated degradation of p53 protein.Citation3 MDM2 when overexpressed creates a genotype similar to one completely lacking p53 expression due to rapid degradation of p53.Citation2 Targeting the various routes through which tumor cells suppress the pro-apoptotic functions of this pathway is now a significant focus of effort in chemotherapeutic agent development.Citation1 While p53 is an inherently unstable protein, mutant p53 is stabilized in part by HSP90 proteins in a manner that affects MDM2 mediated turnover.Citation3
The p53 protein exists in a family consisting of p53, p63, and p73.Citation4 Beginning with the discovery of p73 in 1997, significant work has been focused on the possibility of restoring the function of p53 through its family member p73. Like p53, p73 is expressed in multiple isoforms exhibiting pro- and anti-apoptotic functions. In general, these isoforms are found in 2 categories based on the presence or absence of the N-terminal transactivation domain. These are designated as TAp73 or ΔNp73, with TAp73 featuring pro-apoptotic effects and ΔNp73 featuring anti-apoptotic effects.Citation4 The p73 isoforms have significant function in the regulation of cell cycle arrest as well as important regulatory action in neural stem cell maintenance, and are known to transactivate many p53 transcriptional targets effectively. The inherent redundancy in the p73 isoforms thus makes it a promising potential mechanism to exploit for restoring the pro-apoptotic wild-type p53 pathway in tumors. p73 shares a 63% homology at the transactivation domain with p53.Citation4 Reactivation of the wild-type p53 pathway has already been shown to lead to tumor stasis and regression.Citation3
In this study, we demonstrate that small molecule CB002 and a related compound can restore p53 pathway signaling.
Results
CB002 restores p53 pathway in colorectal cancer cells
To identify p53-restoring small molecules, we screened 50000 small molecular compounds of Chembridge library using a functional cell-based assay in SW480 cells which carry a p53-luc reporter. One compound CB002 (ID is 7745998, IPUA name is 8-anilino-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione) was found to increase p53 responsive bioluminescence( and ). We also identified a CB002-related compound R1 (ID is 849102, IPUA name is 8-anilino-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione) in Chembridge library (). Similar to CB002, R1 increased p53 responsive bioluminescence in a dose dependent manner in SW480 cells ( and ).
Figure 1. Screening experimental compounds CB002 and a related compound R1 for transcriptional activation by p53 in cancer cells. A. Molecular structure of CB002 and R1. B. SW480 with p53 reporter cells were treated with CB002 and R1 in doses ranging from 0 to 20 µmol/L. p53 responsive bioluminescence was imagined by IVIS. Data are representative of triplicate wells. C. The relative increase of bioluminescence in (B). D. Colorectal cancer SW480, DLD-1, HCT116 and HCT116 p53-null cells were treated with CB002 and R1 for 2 and 24 hour. p53 responsive bioluminescence was imagined by IVIS. E. the relative increased bioluminescence in (D) at 2 hours. F. the relative increased bioluminescence in (D) at 24 hours.
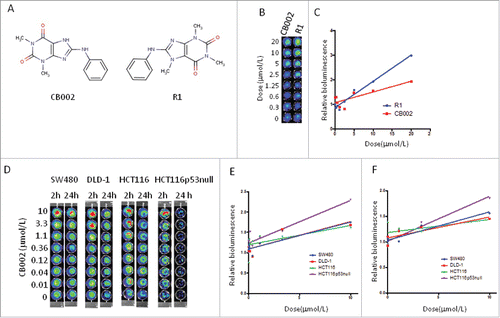
CB002 and R1 were further applied to 4 colorectal cancer cell lines consisting of SW480, DLD1, HCT116, and HCT116 p53 null. As shown in , CB002 increased p53 responsive bioluminescence in both SW480 (mutant p53 R273H, P309S) and DLD-1(mutant p53 S241F) cells in a dose-dependent manner at 2 and 24 hours, as well as in HCT116 p53 null cells ( and ).
For verification of bioluminescence data, western blot analysis of endogenous protein levels of p21, PUMA and DR5 (as representative p53 targets) was conducted by varying drug dose and a time course experiment. In DLD-1 cells (), a minor increase in p21 was observed at 8 hours in both CB002 and R1 at doses of 25 µmol/L and 50 µmol/L. At 24 hours there was a moderate increase in p21 with the moderate dose of 12 µmol/L of CB002 and 25 µmol/L of R1. A minor increase in PUMA was observed at the 25 µmol/L and 50 µmol/L doses for CB002, and an increase at the 25 µmol/L but not the 50 µmol/L of R1 at 8 hours. At 24 hours, an increase in PUMA was observed with exposure to CB002 and R1. DR5 was slightly increased in cells treated with CB002 at 8 and 24 hours, but no change of DR5 was observed in cells treated with R1 in DLD-1 cells.
Figure 2. Effect of CB002 and R1 on p53 pathway signaling in cancer cells. DLD-1 and DLD-p73 Knockdown cells (A), HCT116 (B), and HCT116 p53-null cells (C) were treated with CB002 and R1 for 8 and 24 hours. Protein levels of p53 target genes were determined by Western blot analysis.
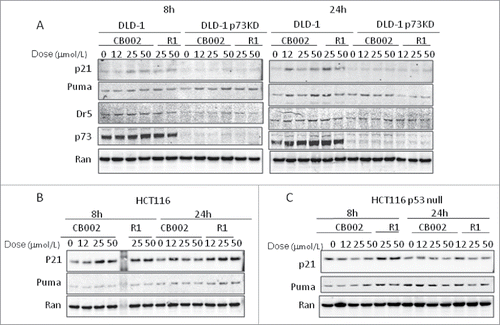
We further applied CB002 and R1 to HCT116 p53-null cells. As shown in , in p53-null HCT116, p21 appeared to increase moderately with R1 and CB002 at 8 hours. At 24 hours no consistent difference was observed for both CB002 and R1 at all dosages with an increase at the single 12 µmol/L dose in compound R1. PUMA appeared to increase at the 50 µmol/L dose at 8 hours and in both the 25 µmol/L and 50 µmol/L doses of R1. At 24 hours no consistent difference in PUMA was observable except a slight decrease in the 2 R1 dosages. On the basis of our data with p53 responsive bioluminescence and p53 target gene expression, we conclude that CB002 and R1 restore p53 pathway responses in DLD-1 and HCT116 p53-null cells.
We also examined the p53 pathway signaling in HCT116 cells which carry with wild-type p53. CB002 increased p53 responsive bioluminescence at 2 hours ( and ). Consistent with the data using bioluminescence, p21 appeared to increase moderately in a dose dependent fashion for both CB002 and R1. PUMA expression appeared to increase only at 24 hours with compound R1 in HCT116 cells (). These data suggest that CB002 reactivates the p53 pathway in wild-type p53 expressing HCT116 cells.
CB002 decreases mutant p53 protein levels in colorectal cancer cells
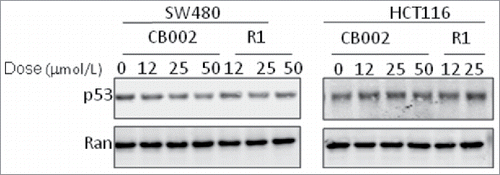
We further examined the effect of CB002 and R1 on mutant p53 protein levels in cancer cells. Mutant p53 protein in SW480 cells and wild-type p53 in HCT116 cells were examined via western blot assay at 24 hours following treatment at varying doses with CB002 and R1. As shown in , mutant p53 protein level appeared to decrease significantly with increasing dose of CB002, and decrease moderately with high dose R1. In contrast, a moderate increase in wild-type p53 was observed with increasing dose of CB002 and R1 in HCT116 cells.
Figure 3. p53 protein level in cancer cells upon CB002 and R1 treatment. SW480 and HCT116 cells were treated with CB002 and R1 for 24 hours. p53 protein was determined by western blotting using anti-p53 (DO-1).
CB002 restores p53 pathway signaling in part through activation of p73 in mutant p53-expressing colorectal cancer cells
CB002 treatment was applied to DLD-1 and p73 knockdown DLD-1 cells at varying doses. Western blot analysis was conducted using DLD-1 and p73 knockdown DLD-1 at 8 hours and 24 hours post-exposure to low ascending doses of CB002 and R1. As shown in , p21, PUMA and DR5 were increased in protein level at 8 and 24 hours in DLD-1 cells treated with CB002 and R1. By contrast, p21 expression was absent for both CB002 and R1 compounds in p73 knock-down DLD-1 cells. PUMA appeared to have increased expression at 8 hours for both compounds over control. At 24 hours, a reduction in PUMA was observed for compound R1 with CB002 largely unchanged from control. DR5 appeared to be unchanged at 8 hours and at 24 hours with less protein level in p73 knockdown DLD-1 cells compared with that in DLD-1 cells. p73 protein level appeared to be unchanged in DLD-1 at 8 hours with a non-dose dependent moderate increase at 24 hours over control. p73 protein was not detected at both time points for both compounds in p73 knockdown DLD-1 cells. These results taken together suggest that knockdown of p73 may have some impact on CB002 and R1-restoring p53 pathway signaling in mutant p53-expressing cancer cells.
CB002 induces cell death in tumor cells with no significant effect on normal cells
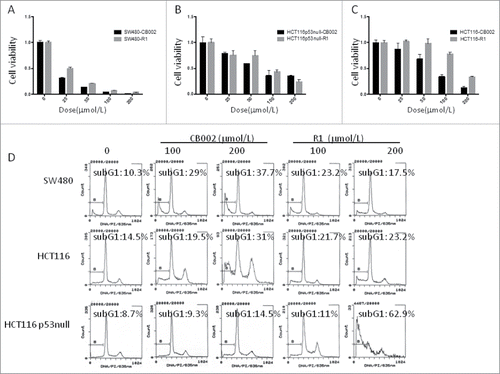
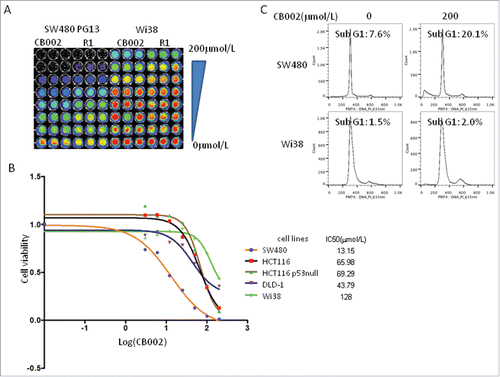
We examined whether CB002 and R1 repress cancer cell growth. To address this issue, cell viability and sub-G1 were determined in colorectal cancer cell lines SW480, HCT116, HCT116 p53−/− upon treatment with CB002 and R1. As shown in , CB002 and R1 both reduced cell viability in SW480, HCT116 and HT116 p53-null cells in a dose dependent manner with CB002 having the greatest effect. Flow cytometry was conducted to assess sub-G1 fraction in SW480, HCT116 and HCT116 p53-null cells at 72 hours post-treatment. Both CB002 and R1 resulted in increased cells in sub-G1 fraction over untreated controls in SW480, HCT116 and HCT116 p53-null cells in a dose dependent manner. CB002 resulted in a greater percentage of cells in sub-G1 fraction than R1 in SW480 and HCT116 at a dose of 200 μM (). These results suggest that CB002 and R1 induce cell death in colorectal cancer cells. CB002 has higher anti-tumor efficacy as compared with R1.
Figure 4. CB002 and R1 induce cell death in colorectal cancer cells. A. Cell viability of SW480 cells treated with CB002 and R1 at 72 hours. B. Cell viability of HCT116 p53-null cells treated with CB002 and R1 at 72 hours. C. Cell viability of HCT116 cells treated with CB002 and R1 at 72 hours. D. Cell cycle profiles of cancer cells SW480, HCT116 and HCT116 p53-null cells. Cells were treated with CB002 and R1 for 72 hrs. Cell viability (A, B, and C) was normalized to DMSO as control. Data are expressed as mean ± SD.
We further applied CB002 to human normal fibroblast Wi38 cells. IC50 of CB002 in Wi38 cells was much higher than those in colorectal cancer cells, SW480, DLD-1, HCT116 and HCT116 p53-null cells ( and ), suggesting that there is a favorable therapeutic index between normal cells and cancer cells. Flow cytometry showed relatively unchanged sub-G1 fraction in normal Wi38 cells treated at the dose (200 μM) of CB002 that effectively increased 20% of cells in sub-G1 in SW480 cancer cells at 72 hours ().
Figure 5. CB002 induces cell death in tumor cells with no significant effect on normal cells. A. Imaging of Cell Titer-Glo as a cell viability assay of SW480 and Wi38 cells treated with CB002 and R1 for 72 hours. B. IC50 of CB002 in cancer cells and normal fibroblast Wi38 cells based on the cell viability. The cells were treated with CB002 for 72 hours. C. Cell cycle profiles of SW480 and Wi38 cells treated with CB002 for 72 hours.
CB002 synergizes with CPT-11 and 5-FU to suppress cell growth in colorectal cancer
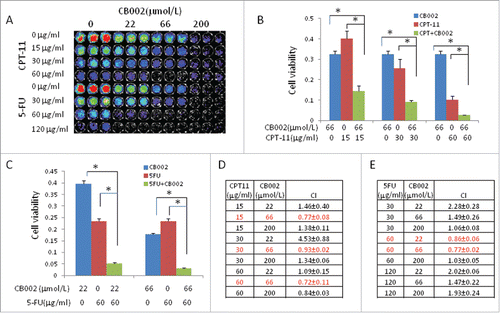
Given that 5-FU and CPT-11 are traditional chemotherapy for colorectal cancer patients often used in combination treatment, we tested whether CB002 can synergize with CPT-11 or 5-FU in inducing cell death in colorectal cancer cells. To address this question, combination treatment of CB002 with CPT-11 or 5-FU was assessed with escalating doses in SW480 cells. As shown in , cell viability of cancer cells had greater reduction with ascending doses of each agent with greater toxicity observed in 5-FU over CPT-11 (). Combination treatment with CB002 (66 μM) and CPT-11 (a series of doses) significantly reduced cell viability in SW480 as compared with the single agent treatments alone (). Further combination index (CI) analysis indicates a synergism of this combination of CB002 and CPT-11 (). Similar to the combination treatment of CB002 and CPT-11, CB002 synergized with 5-FU-induced cell death. As shown in and , combinational treatment of CB002 (22 μM and 66 μM) with 5-FU (60 μg/ml) significantly reduced cell viability in SW480 cells as compared with the single agent treatment alone. Combinational index (CI < 1.0) indicates a synergism of CB002 and 5-FU in cancer cells.
Figure 6. Synergistic effects of CB002 and CPT-11 or 5-FU in treated cancer cells. SW480 cells were treated with CB002 and CPT-11 or 5-FU for 72 hours. A. Imaging of CellTiter-Glo cell viability of SW480 treated with CB002 and CPT-11 or 5-FU. B. Cell viability of SW480 cells treated with CB002 and CPT-11. C. Cell viability of SW480 cells treated with CB002 and 5-FU. D. Combination Index of CB002 and CPT-11. E. Combination Index of CB002 and 5-FU. Cell viability was normalized to DMSO as control. *p < 0.05. Combination index (CI)<1, = 1 and >1 indicate synergism, additive effect and antagonism in drug combination treatment.
Discussion
Targeting the p53 pathway is an attractive strategy for cancer therapy. Small molecular compounds have the potential to restore p53 pathway signaling in cancer cells. Here, we report that small-molecule CB002 and a related compound R1 restore p53 pathway signaling in cancer cells. CB002 and R1 suppress colon adenocarcinoma cell growth with only limited toxicity to normal cell Wi38. There is synergistic efficacy of CB002 with traditional chemotherapeutics CPT-11 and 5-FU. We postulated that CB002 and related compound R1s efficacy in arresting tumor cell growth would be via restoration of the p53 pathway.
We demonstrated that CB002 and R1 are small molecules which successfully increase p53 pathway signaling in cancer cells regardless of p53 status, on the basis of our findings that both compounds not only increased p53 responsive bioluminescence, but also increased the endogenous proteins p21, PUMA and DR5, targets of the p53 pathway in cancer cells ( and ). p53 pathway signaling was increased in mutant p53-expressing DLD-1 and SW480 cells as well as in HCT116 p53-null cells when exposed to CB002 and R1 ( and ). Wild-type p53 is deficient in DLD-1, SW480 cells (which harbor mutant p53) and HCT116 p53-null cells. These results suggest that CB002 increases p53 pathway signaling independent of wild-type p53. p73 is a member of p53 family. We found that knockdown of p73 partially inhibited CB002 and R1-induced p53 pathway signaling (), suggesting that activation of p73 pathway signaling may have some involvement with CB002 and R1-mediated p53 pathway signaling in p53-deficient cancer cells. p73 activation is an alternate pathway contributing to CB002s restoration of p53 pathway signaling. It has been reported that mutant p53 inhibits p73 activation by binding with p73 protein.Citation3 The decrease of mutant p53 may partially contribute to CB002-induced p73 activation. Further study will elucidate how CB002 and R1 activate p73 and how CB002 restores p53 pathway signaling in mutant p53-expressing cancer cells. We also noted that PUMA and DR5 were detected in p73 knockdown DLD-1 cells, although DR5 protein level was reduced in p73-knockdown DLD-1 compared with DLD-1 cells (). Multiple signal pathways are involved in regulating PUMA and DR5 expression in cells.Citation5-8 Our findings suggest that other signal pathways, in addition to p53 family pathway, are involved in response to CB002 and R1 treatment in cancer cells.
It is of note that CB002 and R1 increase p53 signaling not only in p53 deficient cancer cells, but also in wild-type p53 cancer cells ( and ). It is unclear if this increase in p53 signaling is dependent on wild-type p53 or via activation of p73 or both in wild-type p53-expressing cancer cells. Consistent with the CB002 and R1 induced-p53 signaling in cancer cells, CB002 and R1 both induced cell death in cancer cells regardless of p53 status ( and ). These data suggest that CB002 and R1 are not specifically targeting mutant p53-expressing cancer cells.
Gain of function (GOF) of mutant p53 contributes to drug resistance and inhibition of apoptosis. Removal of GOF by targeting mutant p53 has been found to contribute to cell death in cancer cells.Citation3 CB002 and R1 not only restore p53 pathway signaling, but also decrease mutant p53 protein level in cancer cells. In mutant p53-expressing cancer cells SW480, mutant p53 protein level was decreased by CB002 and R1 (). In contrast to mutant p53, wild-type p53 was not decreased in HCT116 cells upon CB002 and R1 treatment. It is known that p53 can be degraded via ubiquitin-proteasome pathway or autophagy.Citation9 Recently, we found a small-molecule NSC59984 that degrades mutant p53 protein in cancer cells.Citation10 The decrease of mutant p53 protein in SW480 cells treated with CB002 might be due to protein degradation. It is also possible that the decrease of mutant p53 by CB002 occurs at the post-translational level. Further study will be undertaken to clarify the mechanism of action of CB002 in decreasing mutant p53 protein level in cancer cells.
CB002 and R1 both induce cancer cell death with limited toxicity to normal cells (). This provides a very favorable therapeutic window, suggesting that CB002 can be used for cancer therapy as a single agent alone. CB002 and R1 both restore or reactivate p53 pathway, but CB002 has higher anti-tumor efficacy compared with R1 (). Combination treatment with novel drugs and traditional chemotherapy is another therapeutic strategy for increasing antitumor efficacy. CB002 was found to have synergy with 5-FU and CPT-11 reducing cell viability in cancer cells (), suggesting that CB002 can be used as combinational treatment in cancer cells. Future in vivo experiments may further evaluate the anti-tumor efficacy of CB002 alone or combination with other agents. Variation was observed between cell lines in response to CB002 ( and ), and further work with larger data sets could clarify CB002s effectiveness in cell lines with suppressed wild-type p53, mutant p53, and the role of p73 activation in CB002s anti-tumor effects. It is important to note that we have not in this manuscript established a role for p73 in the anti-tumor effect of CB002 or R1.
Taken together our results suggest that CB002 and a related compound R1 activate p53 pathway signaling, decrease mutant p53 protein level, and induce cell apoptosis without significant harm to normal cell lines with functioning wild type p53. Gene expression of p53 pathway targets is activated by CB002 and R1. CB002 and related compound R1 are promising therapies for p53-mediated epithelial tumors.
Materials and methods
Bioluminescence assay
Cell-based screening of p53 transcriptional activity for small molecule CB002 was accomplished using noninvasive bioluminescence imaging in human colorectal cancer cell lines SW480, DLD-1, DLD-1 p73−/−, HCT116, and HCT116 p53−/−. These cell lines stably express a p53 reporter, PG13-luc. Cells were seeded in opaque 96-well culture at a density of 5 × 104 cells/well. The cells were treated with CB002 at ranging doses with DMSO controls. Bioluminescence in cells was imaged for p53 transcriptional activity at 2h and 24h using IVIS imaging system (Xenogen).
Cell Titer-Glo luminescent cell viability assay
Cell lines at a concentration of 4 × 103 cells/well were seeded out on an opaque 96-well plate and treated with CB002 and related compound R1 in ranging doses starting from 200 μmol/L with DMSO controls. At 72h after treatment, cells were mixed with 30 μL Cell Titer-Glo reagent and after 10 minutes of room temperature incubation were imaged using IVIS imaging system (Xenogen).
FACS assay
Cells were seeded out at 1 × 106 cells/well on 6 well plates and treated with CB002 and related compound R1 at ranging doses with DMSO controls. Cells were harvested after 72 hours of treatment, all cells including floating cells were fixed with ethanol and stained with Propidium Iodide and then analyzed using Epics Elite flow cytometer to measure the DNA content of the stained cells.
Western immunoblot analysis
Proteins were isolated using NP40 Lysis Buffer [20 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 5 mmol/L EDTA, 50 mmol/L NaF, 1 mmol/L glycerophosphate, 5 mmol/L Na4P2O7, 0.5% NP40, and complete protease inhibitor cocktail (Roche)] and electrophoresed through 4–12% SDS-PAGE followed by semi-dry transfer to PVDF membranes. The PVDF membranes were incubated with different antibodies including p21 (OP64–100UG, EMD Millipore. http://www.emdmillipore.com/US/en/product/Anti-p21WAF1-(Ab-1)-Mouse-mAb-(EA10),EMD_BIO-OP64), PUMA (12450S, Cell Signaling Technology, https://www.cellsignal.com/products/primary-antibodies/puma-d30c10-rabbit-mab/12450), DR5 (3696S, Cell Signaling Technology, https://www.cellsignal.com/products/primary-antibodies/dr5-antibody/3696?N=4294956287&Ntt=3696sandfromPage=plpand_requestid=541668), p53(sc-126, Santa Cruz, https://www.scbt.com/scbt/fr/product/p53-antibody-do-1), and RAN (610341, BD Transduction Laboratories, https://www.bdbiosciences.com/us/reagents/research/antibodies-buffers/cell-biology-reagents/cell-biology-antibodies/purified-mouse-anti-ran-20ran/p/610341) in blocking buffer at 4°C overnight. Bound antibody will be detected using IRDye secondary antibodies (LI-COR Biosciences,) in Odyssey blocking buffer for 1 hour then imaged using the ODYSSEY infrared imaging system.
Disclosure of potential conflicts of interest
W.S.E-D. is a Founder of p53-Therapeutics, Inc., a biotech company focused on developing small molecule anti-cancer therapies targeting mutant p53. Dr. El-Deiry has disclosed his relationship with p53-Therapeutics and potential conflict of interest to his academic institution/employer and is fully compliant with NIH policies and institutional policies regarding this potential conflict of interest.
Funding
This work was supported, in part, by NIH Grant N01-CN43302-WA-17 and N01-CN43302-WA-27. W.S. El-Deiry is an American Cancer Society Research Professor.
References
- Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci USA. 2006;103(29):11003-8. https://doi.org/10.1073/pnas.0604507103. PMID:16835297.
- Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, Moll UM. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9(5):577-88. https://doi.org/10.1158/1541-7786.MCR-10-0534. PMID:21478269.
- Freed-pastor WA, Prives C. Mutant p53: One name, many proteins. Genes Dev. 2012;26(12):1268-86. https://doi.org/10.1101/gad.190678.112. PMID:22713868.
- Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012;3:e285. https://doi.org/10.1038/cddis.2012.27. PMID:22419114.
- Ding WX, Ni HM, Chen X, Yu J, Zhang L, Yin XM. A coordinated action of Bax, PUMA, and p53 promotes MG132-induced mitochondria activation and apoptosis in colon cancer cells. Mol Cancer Ther. 2007;6(3):1062-9. https://doi.org/10.1158/1535-7163.MCT-06-0541. PMID:17363499.
- He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene. 2004;23(14):2554-8. https://doi.org/10.1038/sj.onc.1207351. PMID:14691451.
- Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7(1):45-52. https://doi.org/10.1016/j.celrep.2014.02.036. PMID:24685133.
- Farooqi AA, Li KT, Fayyaz S, Chang YT, Ismail M, Liaw CC, Yuan SS, Tang JY, Chang HW. Anticancer drugs for the modulation of endoplasmic reticulum stress and oxidative stress. Tumour Biol. 2015;36(8):5743-5752. https://doi.org/10.1007/s13277-015-3797-0. PMID:26188905.
- Muller PA, Vousden KH, Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell. 2014;25(3):304-317. https://doi.org/10.1016/j.ccr.2014.01.021. PMID:24651012.
- Zhang S, Zhou L,Hong B, van den Heuvel AP, Prabhu VV, Warfel NA, Kline CL, Dicker DT, Kopelovich L, El-Deiry WS. Small molecule NSC59984 restores p53 pathway signaling and anti-tumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Cancer Res. 2015;75(18):3842-52. https://doi.org/10.1158/0008-5472.CAN-13-1079. PMID:26294215.