
ABSTRACT
The emergence of haematopoietic stem and progenitor cells (HSPCs) from hemogenic endothelium results in the formation of sizeable HSPC clusters attached to the vascular wall. We evaluate the cell cycle and proliferation of HSPCs involved in cluster formation, as well as the molecular signatures from their initial appearance to the point when cluster cells are capable of adult engraftment (definitive HSCs). We uncover a non-clonal origin of HSPC clusters with differing cell cycle, migration, and cell signaling attributes. In addition, we find that the complement cascade is highly enriched in mature HSPC clusters, possibly delineating a new role for this pathway in engraftment.
Introduction
Hemogenic endothelium (HE) exists during a narrow window (∼E9.5 to E11.5) in development.Citation1-7 During this period, these specialized endothelial cells have the ability to generate the first definitive haematopoietic stem and progenitor cells (HSPCs) in the embryo. Hemogenic vascular beds are contained intra-embryonically in the aorta-gonad mesonephros region (AGM)Citation1,2,8-10 as well as extra-embryonically in the placenta,Citation11,12 yolk-sac,Citation13-16 and umbilical and vitelline arteries.Citation10,17,18 Hemogenic vascular beds are uniquely characterized by the presence of haematopoietic cell clusters, a distinct group of haematopoietic cells that bud off of the endothelial wall.Citation1,4,10
The dorsal aorta is the most extensively studied hemogenic vascular bed, and is noted to contain several clusters of haematopoietic cells termed intra-aortic haematopoietic clusters (IAHCs).Citation1,2,19,20 These structures have been observed to be as large as ∼30 cells and are thought to initially form from a Runx1 dependent endothelial-to-haematopoietic transition (EHT),Citation5,19,21,22 and contain both haematopoietic stem and progenitor cells (hence HSPCs). Cells comprising IAHCs can be identified by surface marker expression of CD31 and CD117, as well as lack of expression of germ line marker SSEA-1.Citation19 Whole embryonic AGM regions have been shown to engraft irradiated adult hostsCitation2,23,24 at E11, but also E10 albeit with very low incidence.Citation24
Further transplantation experiments of isolated IAHC cells have been performed with sorted SSEA-1−/CD31+/CD117+ cells (IAHC populations) from E11.5 AGMs and shown to engraft irradiated adults.Citation19 Thus, the E11 IAHC cell population contains both definitive HSCs and progenitor cells. However, transplantation of the same sorted IAHC cell populations from E10.5 AGMs shows undetectable repopulation activity in adult irradiated mice,Citation19,24,25 but does demonstrate engraftment in neonates.Citation20 The neonatal engraftment at E10.5 suggests that the IAHCs also contain haematopoietic stem and progenitor cells, but that the HSC population is immature or a pre-HSC population.Citation20,23 Overall, these data demonstrate that IAHC populations may only possess adult engraftment potential later in the HE time window, suggesting a maturation process from E10 to E11 that allows for adult engraftment.Citation20,25,26 Thus, during this transient period in development, control of pre- HSC maturation may be spatially and temporally regulated. Understanding the establishment of this multi-step maturation process is critical to efforts of generating HSCs in vitro for clinical applications. To begin to understand IAHC generation and subsequent maturation steps, we evaluated IAHC cell proliferative capacity, cell cycle attributes, clonal origins from HE cells, as well as possible critical signaling pathways. Here we propose that the pre-HSC maturation process includes changes in cell cycle dynamics and activation of the complement system.
Results
Analysis of IAHC cell cycle kinetics reveals a proliferative population following HE emergence
We set out to determine how newly emerged haematopoietic cells form a cluster. To address this, we first defined an IAHC as a group of 3 or more haematopoietic cells attached to the endothelium. To identify IAHCs, we used PU.1 as a nuclear marker of IAHCs during E10.5 and E11.527 (Fig. S1A–C). To determine IAHC size and distribution, we examined the frequency of IAHCs according their size (cell number) at E10.5, the time point that corresponds to the peak of IAHC formation.Citation19 Quantification of IAHCs at E10.5 revealed a wide distribution of IAHC size that ranged from IAHCs with 3 cells to 47 cells (Fig. S1D). Interestingly, at E11.5 the distribution range of IAHC size narrowed from IAHCs with 3 cells to 11 cells (Fig. S1E). Overall, the data suggest that most IAHCs form at E10.5, the time point thought to be peak of IAHC formation.Citation19
To understand how some IAHCs could contain >20 cells, we next determined the extent cell proliferation contributed to the large number of cells within each IAHC. To determine if IAHCs are proliferative, we injected the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU) in pregnant females at E10.5 and separately at E11.5, and evaluated the embryos 2 hours later (). Immunofluorescence for EdU distinguishes cells in the S-phase from cells in other phases of the cell cycle,Citation28,29 and Ki67 labels cells in all active phases of the cell cycle (except G0).Citation30 Our analysis reveals several cells in the IAHCs display markers of proliferation ( and ). Moreover, quantification of IAHCs that express these markers show that ∼90% of cells in IAHCs were Ki67+, and ∼42% of these cycling IAHC cells were EdU+ ( and ) at E10.5. These percentages were similar upon examination of IAHCs at E11.5 ( and ). Together, these data show that these 2 time points in the hemogenic window contain cells within IAHCs that are proliferative.
Figure 1. IAHCs are proliferative and their cell cycle length increases during the later stage of the hemogenic window. (A) EdU labeling experimental schema. Pregnant wildtype animals at E10.5 and E11.5 were injected intraperitoneally (IP) with 1.5 mg of EdU 2 hours before evaluation of embryos. (B) Immunofluorescence of proliferation markers, EdU an S-phase marker (green) and Ki67, a general proliferation marker (expect G0) (gray) in the dorsal aorta at E10.5. CD31 (red) delineates the endothelium. The white dotted box outlines an IAHC. Scale bar, 20 µm. (C) Higher magnification of the E10.5 IAHC in (B) containing labeled EdU+ and Ki67+ cells. CD31 marks the endothelium and IAHCs. Scale bar, 10 µm. (D) Quantification of Ki67+ cells in IAHCs expressed as percentage of total cells. (E10.5 mean = 90.1% vs E11.5 mean = 83.46% p = 0.50) (E) Quantification of EdU+ cells in IAHCs expressed as the percentage of the total number of Ki67+ cells. (E10.5 mean = 45.2% vs E11.5 mean = 27.9% p = 0.081) A total of n = 47 IAHCs were analyzed at E10.5 and a total of n = 44 IAHCs were analyzed at E11.5. (F) EdU and BrdU sequential labeling experimental schema. Pregnant wildtype animals at E10.5 and E11.5 were sequentially pulsed with EdU followed by BrdU. 1.5 hours apart. This technique allows for identification of cells that are in the S-phase (S) (EdU+BrdU+) and cells leaving the S-phase (L) (EdU+BrDU−). All proliferating IAHC cells are marked by PU.1 (P). The total length of S phase is estimated by first calculating the number of cells leaving the S-phase (L) over the total number of cells in the S-phase (S). The initial time (Ti) between EdU and BrdU (1.5 hrs) is then divided by this fraction, Ts = Ti/(L/S). The total length of the cell cycle (Tc) is estimated by first determining the number of cells in the S-phase over the total number of proliferating cells (P). The total S-phase length (Ts) is then divided by this fraction, Tc = Ts/(S/P). (G) Immunofluorescence of an IAHC containing EdU+ (green) and BrdU+ (red) cells. PU.1 (gray) was used to distinguish IAHCs. This representative image depicts an IAHC that contains cells at different phases of the cell cycle given their varying incorporation of EdU (green), BrdU (red) or both (yellow). A total of 81 IAHCs were evaluated at E10.5 and a total of 87 IAHCs were evaluated at E11.5. Scale bar, 10 μm. (H) Comparison of the proportion of cells in the S phase (EdU+BrdU+, yellow) vs. other cell cycle phases during E10.5 and E11.5 (Fisher's exact test, p < 0.001). At E10.5 most IAHCs contain cells that are in S phase (56% S-phase vs. 44% other phases) while at E11.5, most IAHCs contain cells in the other phases of cell cycle (32% S-phase vs. 68% other phases). (I) Comparison of the S-phase length between E10.5 and E11.5 shows no difference (0.78 h vs 0.74 h, t-test with Welch's correction p < 0.9). Comparison of the total cell cycle length between E10.5 and E11.5 demonstrates lengthening of the cell cycle (5.38h vs 8.16 h, t-test with Welch's correction p < 0.003). All data shown as mean +/− SD. All experiments were conducted with n = 2–3 Ls.
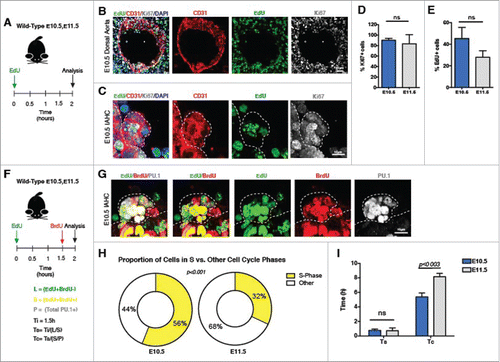
Since IAHCs contain proliferating cells, we next determined their average generation time (cell cycle length). We labeled IAHCs with a sequential pulse of nucleoside analogs, 5-ethynyl-2′-deoxyuridine (EdU) followed by 5-bromo-2′-deoxyuridine (BrdU). The sequential administration of these compounds allows for the determination of the cell cycle kinetics of proliferating IAHC cells by separating cells into early (EdU−BrdU+), mid (EdU+BrdU+), and late (EdU+BrdU−) S phasesCitation31,32 (). To validate the system, we evaluated EdU and BrdU in the neural tube at E10.5 and noted comparable numbers to a previous reportCitation31 (Fig. S2A–C). To adapt the system for IAHC evaluation, we used PU.1 as a nuclear marker of IAHC cells, which we have previously determined to contain about ∼90% Ki67+ cells, as an estimate of the total number of proliferating cells ( and Fig.S1A–C). Analysis of the dual pulse labeling revealed that a proportion of the cells within IAHCs at E10.5 are actively in the S-phase (versus 42% by Ki67/EdU staining, ), while a proportion of cells within IAHCs at E11.5 are positioned in other phases of the cell cycle (, E10.5 vs. E11.5, p < 0.001). Furthermore, we estimated the total length of the cell cycle (Tc) of IAHC cells at E10.5 to be 5.38 hours and the S-phase length (Ts) at 0.78 hours ( and ). To gain further insight into the cell cycle kinetics of IAHC cells, we extended this analysis to E11.5 and found that the Tc lengthens to 8.16 hours (, Tc E10.5 vs E11.5, p < 0.003), while Ts remains the same (, Ts E10.5 0.78 h vs. E11.5 0.74 h). Overall, these data suggest that IAHC cells are capable of dividing 4.5 times within the 24 hour period of time leading to the peak of the hemogenic window (E10.5), such that a single hemogenic endothelial clone cycling at the peak rate could engender up to 22.6 cells or ∼23 cells (). Furthermore, these observations also suggest that cell division alone from a single hemogenic clone could not account for IAHCs with greater than 23 cells in a 24-hour period.
Table 1. Estimation of cell number from single clonal divisions at E10.5 and E11.5.
Clonal analysis of haematopoietic clusters arising from hemogenic endothelium
We next explored through low frequency labeling/fate tracing of endothelial cells whether IAHCs are derived from one or more hemogenic endothelial clones.Citation33,34 To investigate, we initially crossed a constitutive VE-cadherin-Cre to the R26r-mTmG reporter line (VECAD/mTmG) (Fig. S3A). Recombination in this line results in the excision of mTomato (mTom) and expression of mGFP in endothelial cells (CD31+CD117−) and their haematopoietic (CD31+CD117+) derivatives, including IAHCs within the embryo (Fig. S3A). Upon quantification, we found mGFP+ labeling in ∼75% of endothelial cells (CD31+CD117−) and ∼50% of IAHC cells (CD31+CD117+; Fig. S3A and B). Immunofluorescence also revealed mosaic expression of mGFP and Tomato in the endothelial cells and IAHC cells in the DA (Fig. S3C and D). Interestingly, 39% of IAHCs contained both mGFP+ and mTom+ cells (Fig. S3E). About 53% of the total IAHCs examined (n = 57) were comprised of mGFP+ cells only, while 9% of IAHCs contained cells that were not Cre recombined, and hence mTom+ only (Fig. S3E). Together, these data indicate that distinct HE clones may contribute to the formation of IAHCs. To test this hypothesis, we performed low dose clonal labeling using Cdh5(PAC)-CreERT crossed to the R26r-mTmG reporter line (iCdh5/mTmG) (Fig. S3F-H). A range of low dose tamoxifen (50–150µg) was administered to pregnant females at E8.5 before any IAHC formation to label only endothelial clones, and IAHCs were evaluated at E10.5, the peak of IAHC formation,Citation19 for percent recombination in endothelial cells and IAHC cells, Fig. S3F-H. The analysis revealed that a dose of 100 µg allowed for ∼10% labeling of the dorsal aorta (ECs and IAHC cells). We then administered the optimal clonal labeling dose of 100µg (0.1mg) at E8.5 and analyzed the clonal labeling of IAHCs at E10.5 (). Outcomes of the experiment are identified as IAHCs from a single origin (mGFP+only), polyclonal origin (mGFP+/mTom+) or unlabeled (no Cre recombination (mTom+only)) (). Polyclonal in this context only designating more than one clone. Following induction, we observed mosaic mGFP+ and mTom+ labeling in the endothelium and cells comprising IAHCs of the DA (, ). For our analysis of clonality, we excluded mTom+ only IAHCs because we could not conclude whether those IAHCs arose from a single clone since no Cre recombination/fate tracing took place. We evaluated 190 IAHCs (n = 5 embryos) and noted 46 IAHCs at E10.5 were either monoclonal (mGFP+) or polyclonal (mGFP+/mTom+). Quantification of single color (mGFP+ only) and mosaic IAHCs (mGFP+ /mTom+) at E10.5 revealed that the majority of IAHCs (41 out of 46) were mosaic (), while only 5 out of 46 IAHCs were from a mGFP+ only clone. We further extended this analysis to IAHCs at E11.5. We evaluated 59 IAHCs and found 14 IAHCs to be either monoclonal (mGFP+) or polyclonal (mGFP+/mTom+), (). At this time point we observed a similar trend as at E10.5 where most IAHCs were mosaic (11 out of 14) and minority derived from a single mGFP+ clone (3 out of 14) (). Together, the data suggest that IAHCs largely derive from more than one clone (polyclonal origin). Next, we wanted to determine whether any difference in clonality existed between small IAHCs (3–9 cells) vs. large IAHCs (≥10 cells). Our analysis revealed that small IAHCs and large IAHCs significantly differed in their clonality, where small IAHCs were more likely to be derived from one clone (, Fisher's Exact = 0.0495). To further support these observations, we also used the confetti reporter system by crossing inducible endothelial Cre animals with the confetti reporter line (). Outcomes of this cross yield random recombination and labeling of endothelial cells and their haematopoietic derivatives that can be identified with 4 distinct fluorescent labels: mCFP, RFP, YFP and GFP (). In terms of inferring clonality, single color IAHCs denote monoclonal origin and multi-color IAHCs denote polyclonal origin (). Induction with 2 mg of tamoxifen resulted in low percentage of recombination (< 5%) which may be due the inefficiency of the iCdh5-Cre, or perhaps that the relatively short cell cycle length of these IAHC cells is precludes detectable accumulation of the fluorescent protein after recombination. Quantification of labeled IAHCs (n = 4) at E10.5 showed that IAHCs contained cells that were labeled with 2 distinct fluorescent colors (, E). Collectively, these data further support that IAHCs largely derive from more than one clone, and likely derive from 2 clones (biclonal vs. polyclonal origin).
Figure 2. Low dose clonal labeling of IAHCs reveal clonal heterogeneity and polyclonal origin. (A) Experimental schema: Cdh5(PAC)-CreERT2 animals were crossed with R26r-mTmG lines. Pregnant females were induced with a low dose of tamoxifen (0.1mg) at E8.5 before IAHC formation. Embryos evaluated at at E10.5 and E11.5. (B) Potential outcomes following low dose induction: unlabeled (no Cre recombination, mTom+ only, monoclonal (mGFP+ only) and polyclonal (mGFP+mTom+)). (C) Fluorescence of the dorsal aorta (DA) at E10.5 (broken lines) after low dose induction shows mosaic mGFP+ (green) expression in a background of mTom+ (red) cells. PU.1 (gray) labels an IAHC. Scale bar, 20 μm. (D) Higher magnification of the IAHC depicted in (C) which is noted to contain both mGFP+ (green) and mTom+ (red) cells. PU.1 (gray) labels cells in the IAHC and some single haematopoietic cells. Scale bar, 10 μm. (E) Analysis of IAHC clonality at E10.5. Monoclonal (mGFP+ only) IAHCs denoted by light blue bars, and polyclonal (mGFP+mTom+) IAHCs denoted by dark blue bars, unlabeled IAHCs denoted by clear bars. A total of 190 IAHCs were analyzed. n = 5 litters. (F) Analysis of IAHC clonality at E11.5. Monoclonal (mGFP+ only) IAHCs denoted by light gray bars, and polyclonal (mGFP+mTom+) IAHCs denoted by dark gray bars, unlabeled IAHCs denoted by clear bars. A total of 59 IAHCs were analyzed, n = 3 litters.
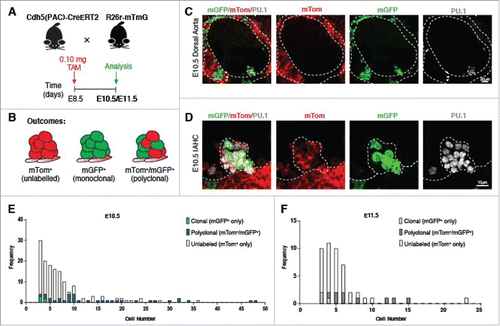
Table 2. Total IAHCs evaluated for clonality at E10.5 and E11.5.
Table 3. Cluster size and clonality at E10.5.
IAHCs are positioned in different phases of the cell cycle as the hemogenic window progresses
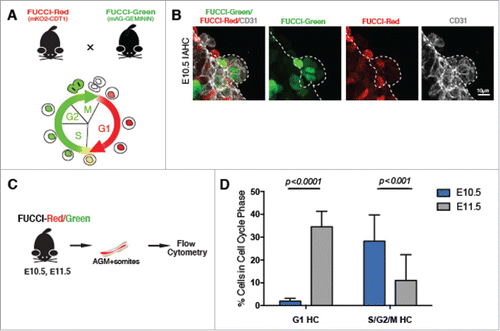
Our finding that a higher proportion of IAHC cells were mainly in the S phase at E10.5 vs. E11.5, together with the increase in the average cell cycle length (–I) prompted us to evaluate whether cells comprising IAHCs may be positioned in different cell cycle phases during early (E10.5) and later (E11.5) periods of the hemogenic window. To test this, we used the FUCCI (Fluorescence Ubiquitin Cell Cycle Indicator) reporter mouse lines.Citation35 In this line, CDT1-mKusabiraOrange2 (mKO2) marks cells in G1 and GEMININ-AzamiGreen (mAG) marks cells in S/G2/M (). We first verified the expression of these markers relative to DAPI incorporation (Fig. S4A) to determine if the fluorescently labeled cells with either mKO2-CDT1 and/or mAG-GEMININ corresponds to appropriate cell cycle stage by DNA content. We then evaluated expression in the developing neural tube and AGM (Fig. S4B-C), and confirmed the pattern of G1 (FUCCI-Red) vs. S/G2/M (FUCCI-Green) in the neural tube. In this region, the somites are labeled with FUCCI-Red denoting that cells are in G1, while the rest of cells in the neural tube are labeled with FUCCI-Green indicating that they are in the S-phase.Citation35 We also evaluated immunofluorescence for the protein GEMININ in the context of the transgenic reporter FUCCI-Green (GEMININ-AzamiGreen (mAG)) within the developing endothelium and IAHC cells (Fig. S4D), and found that the mAG reporter tends to underreport (by 12%) cells that express the protein GEMININ (via immunofluorescence), Fig. S4E. With that in mind, we evaluated cells within IAHCs for their expression of FUCCI-Green and FUCCI-Red, and noted that cells which make up a single IAHC are positioned in different cell cycle phases. Thus, the cells comprising one IAHC are not necessarily synchronized in their cell cycles (). To analyze the IAHC cell population as a whole, we used FACS to discern the cell cycle phase of IAHC cells at E10.5 and E11.5. FACs analysis revealed that at E10.5 ∼2% of the SSEA-1−CD31+CD117+mKO2+ IAHC cells population (IAHCs) resides in G1, while ∼28% resides in the S/G2/M ( and ). Interestingly, at E11.5 ∼35% of the IAHC cell population now resides in G1, and ∼11% resides in S/G2/M ( and ). Overall, the data suggest that there is a shift in the cell cycle position of cells within the IAHCs as the hemogenic window progresses during development. This previously unappreciated difference between the cell cycle position of IAHC cells at E10.5 and E11.5 have led us to postulate whether the cell cycle position may influence repopulation potential.
Figure 3. IAHCs are positioned at different phase of the cell cycle during E10.5 and E11.5. (A) FUCCI-Red (mKO2-CDT1) were crossed with FUCCI-Green (mAG-GEMININ) animals. In G1, active CDT1 tagged with Kusabira Orange (mKO2) is present thereby marking cells in G1. In S/G2/M, GEMININ is tagged with Azami Green (mAG) abeling cells in S/G2/M, while CDT1 is degraded (along with mKO2). (B) Immunofluorescence of an IAHC at E10.5 shows cells in G1 labeled by mKO2 (red) and cells in S/G2/M (green) labeled by mAG. Broken lines depict the IAHC. CD31 (gray) marks the endothelium. Scale bar, 10 μm. (C) FUCCI AGMs at E10.5 and E11.5 were analyzed by flow cytometry for cell populations in G1 and S/G2/M by their respective fluorescence mKO (red) and mAG (green), (D) Comparison of IAHC populations (DAPI−SSEA1−CD31+CD117+) in G1 and S/G2/M at E10.5 (blue bars) and E11.5 (gray bars). The data demonstrate that at E10.5 the majority of IAHC cells are in S/G2/M vs. G1 (28.2% S/G2/M vs. 1.92% G1, t-test with Welch's correction p < 0.0001) while at E11.5, majority of the IAHC cells are G1 v. S/G2/M (11% S/G2/M vs. 34.5% G1, t-test with Welch's correction p < 0.001). n = 4, All data shown as mean +/− SD.
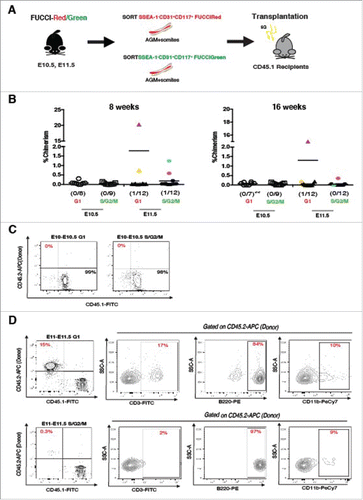
Cell cycle phase does not fully account for repopulation activity
Previous studies from Nygren and colleagues which examined the cell cycle kinetics of E14.5 fetal-liver HSCs (FL-HSCs) found that a prolonged cell cycle length is a characteristic of HSCs.Citation36 In line with these observations, our data demonstrate a prolongation of the cell cycle length at E11.5 which is coincident with the observed repopulation activity during this time point.Citation8,19,23,24,37,38 To determine whether cell cycle position might influence the behavior of IAHC cells in a reconstitution assay, we directly transplanted irradiated adult animals with FACS sorted SSEA-1−CD31+CD117+mKO2+ cells (G1 IAHCs) and SSEA-1−CD31+CD117+mAG+ cells (S/G2/M IAHCs) from E10.5 and E11.5 AGMs (). We find that regardless of their phase in the cell cycle, E10.5 IAHC cells (n = 8–9 per group) have undetectable levels of chimerism even after 16 weeks post-transplantation ( and ). Conversely, only E11.5 IAHC cells showed engraftment (≥1% donor-derived cells) within 8 weeks and 16 weeks. Yet, some animals demonstrate only transient levels of engraftment (). Of the E11.5 transplanted animals (n = 12 each group) we determined to have been repopulated by 8 weeks (1 each in of the G1 and S/G2/M groups respectively), only one had stable engraftment by 16 weeks as measured by peripheral blood chimerism (). In the 16-week engraftment cohort, the animal transplanted with G1 IAHC cells demonstrated stable repopulation while the animal transplanted with S/G2/M IAHC cells did not appear to maintain the >1% level of engraftment long-term (, G1 IAHCs 14.5% vs S/G2/M IAHCs 0.3%). Further evaluation of lineage markers for B and T cells as well as myeloid lineage cells within these animals revealed that these cells were indeed able to reconstitute the different haematopoietic cell lineages (). Overall, the data suggest that enrichment of E10.5 IAHC cells in G1 does not overcome their inability to engraft adult hosts, as neither does enrichment for cells in S/G2/M. However, our data also support that E11.5 IAHCs contain adult engraftable HSCs (especially considering the low numbers of cells transplanted), and that the E11.5 HSC population may be enriched when selected for the G1 (vs. S/G2/M) cell cycle phase, similar to their fetal and adult HSC counterparts.Citation36,39-41
Figure 4. Direct transplantation of E10.5 and E11.5 FUCCI labeled IAHCs. (A) Transplantation experimental scheme. IAHC cell populations (DAPI−SSEA1−CD31+CD117+) from FUCCI-Red (mKO2-CDT1) and FUCCI-Green (mAG-GEMININ) mice were sorted (cell number = 200–2000 cells per group) and directly transplanted into irradiated adult recipients (E10.5 n = 8–9 per group, E11.5 n = 12 per group). (B) Analysis of engraftment (defined as >1% peripheral blood chimerism) at 8 and 16 weeks depicts E10.5 FUCCI-Red (open circles) and green (open squares) IAHCs with no detectable engraftment (0/8 and 0/9 respectively). E11.5 FUCCI-Red (triangles) and FUCCI-green (hexagons) each exhibit one animal with detectable levels of engraftment (1/12 and 1/12 respectively) at 8 weeks (each labeled with a separate color). Only the recipient of G1 IAHCs retains stable engraftment at 16 weeks (fuchsia triangle). No stable engraftment was seen in E11.5 Fucci-Green at 16 weeks, although there was low level of chimerism detected (0.3%) (fuschia hexagon). Double asterisks indicates the occurrence of mortality in the cohort. (C) Representative FACS plots of E10.5 FUCCI-Red and FUCCI-Green IAHCs show no detectable engraftment by 16 weeks. (D) FACS plots of E11.5 G1 IAHCs (fuschia triangle) demonstrate 15% donor reconstitution and peripheral blood contribution to the T (CD3-FITC), B (B220-PE) and myeloid (CD11b-PeCy7) lineages by 16 weeks and for E11.5 S/G2/M (blue hexagon) 0.3% donor engraftment and lineage contribution to B, T, and myeloid lineages.
Cell cycle-specific transcriptional profiling of E10.5 and E11.5 IAHC cells reveal transcriptional programs related to cell movement and cell migration
To further define transcriptional differences that underlie the developmental age and maturation of cells comprising the IAHCs, we performed RNA sequencing on 4 samples: E10.5 and E11.5 SSEA-1−CD31+CD117+mKO2+ (G1 IAHCs) and E10.5 and E11.5 SSEA-1−CD31+CD117+mAG+ (S/G2/M IAHCs) (). We have identified a total of 774 genes for G1 and 703 genes for S/G2/M that are differentially expressed between E10.5 and E11.5. G1 contained 453 unique genes and S/G2/M contained 381 unique genes (). To determine the molecular signature of IAHCs that change according to developmental age, only genes with a 2-fold change in expression were analyzed using the Ingenuity Pathway Analysis (IPA, Ingenuity Systems, www.ingenuity.com). We used 2 metrics, the p-value and the activation z-score, to identify the most important downstream effects related to age. A positive z-score indicates an increased functional or pathway activity in E11.5 relative E10.5 for both G1 and S/G2/M samples. The p-value, calculated with Fisher's exact test, indicates the likelihood that the association between a set of genes in our data set and a biologic function or pathway is significant.
Figure 5. Transcriptional profile of E10.5 and E11.5 FUCCI labeled IAHCs. (A) RNA sequencing experimental scheme. IAHC cell populations (DAPI−SSEA1−CD31+CD117+) from FUCCI-Red (mKO2-CDT1) and FUCCI-Green (mAG-GEMININ) mice were sorted and subjected to RNA sequencing. (cell number, n = 300 – 8000 cells pooled from 2–3 litters per group per age). (B) Venn diagram depicts the number of common and unique genes between G1 (FUCCI-Red) and S/G2/M (FUCCI-Green) IAHCs. We identified 453 unique genes upregulated in G1 E11.5 IAHCs vs. G1 E10.5 IAHCs. Similarly, we identified 381 unique genes upregulated in S/G2/M E11.5 IAHCs vs. S/G2/M E10.5 IAHCs. These 2 groups share 321 common genes. (C) The RNASeq analysis of top biologic functions (z-score >5, p < 0.05) enriched in E11.5 IAHCs at G1 relative to E10.5 IAHCs at G1 identifies the top 11 increased biologic functions involved a variety of cellular processes including: phagocytosis, organization, migration, immune response, homing, endocytosis, differentiation, chemotaxis, cell viability, cell survival and cell movement. z-score ≥ 5 (black dotted line), p-value (red dots) via Fisher's exact test. (D) The RNASeq analysis of top biologic functions (z-score >5, p < 0.05) enriched in E11.5 IAHCs at S/G2/M relative to E10.5 at S/G2/M reveals the top 6 increased biologic functions that are involved in: cell survival, migration, homing, development, cell viability and cell movement. Not surprisingly, these categories are similar to those in E11.5 IAHCs at G1. z-score ≥ 5 (black dotted line), p-value (red dots) via Fisher's exact test. (E) The analysis of canonical pathways (z-score >5, p < 0.05) that are active in E11.5 IAHCs at G1 compared with E10.5 IAHCs at G1 identifies the top 10 activated signaling pathways which include: Gamma receptor-mediated phagocytosis, production of NO, leukocyte extravasation, IL-8 signaling, Rac signaling, integrin signaling, dendritic cell maturation, NFAT signaling, NF-Kappa B singaling and Rho GTPase signaling. (F) The analysis of canonical pathways (z-score >5, p < 0.05) that are active in E11.5 IAHCs at S/G2/M compared with E10.5 IAHCs at S/G2/M identifies the top 10 activated signaling pathways which include: Gamma receptor-mediated phagocytosis, ERK/MAPK signaling, production of NO, PKC delta signaling, leukocyte extravasation, NFAT signaling, p38 MAPK signaling, Tec Kinase signaling, IL-8 signaling and integrin signaling.
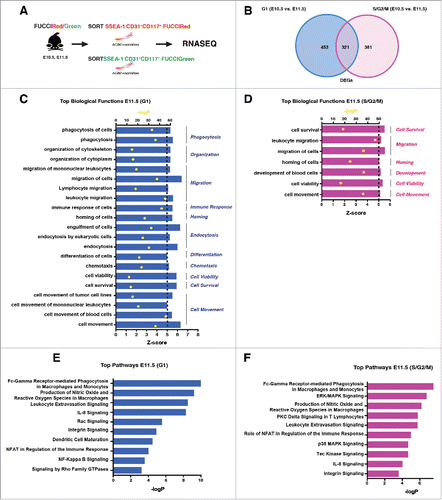
Our analysis revealed that genes overexpressed in E11.5 vs. E10.5 at G1, regulate 11 top functions related to variety of cellular processes (). Interestingly, the functions that are significantly increased are those that are involved in cell migration, endocytosis, cell movement, phagocytosis, cell organization, immune response, homing differentiation and cell survival. Perhaps not surprising is that the analysis of genes overexpressed in E11.5 vs. E10.5 at S/G2/M, yielded 6 top functions similar to those in G1 with cell migration, cell movement, homing, cell survival, and development being significantly activated (). Furthermore, examination of top canonical pathways in G1 reveal activation of signaling that regulate phagocytosis, nitric oxide signaling, leukocyte extravasation and IL-8 (). Our analysis of canonical pathways in S/G2/M show activation of phagocytosis, ERK signaling, nitric oxide signaling and PKC Delta signaling (). Overall, our data suggest that regardless of the cell-cycle phase, a cell movement and cell migration program is established in IAHC cells by E11.5.
Molecular differences between E11.5 G1 and S/G2/M IAHC cells point to complement activation for a role in engraftment
Since our transplantation data suggest that both E11.5 G1 and S/G2/M IAHCs contain cells with different engraftment potential, we wanted to further determine molecular differences related to the cell cycle that may influence engraftment. We identified 106 differentially expressed genes between G1 and S/G2/M IAHCs at E11.5. First, we compared transcripts from E11.5 S/G2/M with E11.5 G1 to identify functions that are upregulated within the G1 phase of the cell cycle. Surprisingly, genes overexpressed in G1 regulate 14 main functions that are involved in different cellular processes. The biologic functions that are most significantly activated include chemotaxis, migration, uptake and cell volume (). As expected, comparison of E11.5 G1 with E11.5 S/G2/M transcripts to identify functions upregulated within the S/G2/M phases of the cell cycle yielded functions relating to proliferation, infection and growth () with proliferation being the most significantly activated.
Figure 6. Molecular differences between E11.5 G1 IAHCs and E11.5 S/G2/M IAHCs. (A) Our analysis of top biologic functions (z > 0, p < 0.05) enriched in E11.5 G1 IAHCs relative to E11.5 S/G2/M IAHCs reveal the top 15 upregulated functions in G1 which include: uptake, synthesis, quantity, proliferation, migration, metabolism, corpuscular volume, invasion, immune response, export, engulfment, endocytosis, differentiation, chemotaxis and viability. (B) Our analysis of top biologic functions (z > 0, p < 0.05) enriched in E11.5 S/G2/M IAHCs relative to E11.5 G1 IAHCs reveal the top 3 functions increased in S/G2/M which are: proliferation, infection and growth. (C) The canonical signaling pathways that are activated (z>0, p < 0.05) in E11.5 G1 IAHCs compared with E11.5 S/G2/M IAHCs include: complement system, pattern recognition receptors, LXR/RXR activation. (D) Complement transcripts are increased in E11.5 G1 IAHCs relative to E11.5 S/G2/M IAHCs. FPKM, Fragments Per Kilobase of transcript per Million. (E) The canonical signaling pathways that are activated (z > 0, p < 0.05) in E11.5 S/G2/M IAHCs compared with E11.5 G1 IAHCs include: G2/M DNA damage checkpoint, mitotic roles for polo-like kinases and melatonin degradation. (F) Cell cycle related genes have increased activation during the S/G2/M phases of the cell cycle regardless of developmental age. FPKM, Fragments Per Kilobase of transcript per Million. (G) QPCR validation of complement-related genes. Genes with significant changes in expression were C1QA, C1QB, CD93, and C3AR (t-test with Welch's correction *p < 0.05 ** p < 0.005), normalized to Rpl3 expression, n = 3. Data expressed as mean +/− SD.
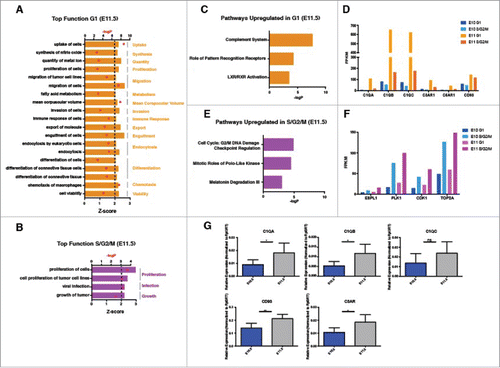
Our analysis of upregulated pathways in E11.5 G1 vs. S/G2/M also revealed activation of canonical signaling pathways that regulate the complement system, roles of pattern recognition receptors and LXR/RXR activation (). Indeed, several complement genes, such as receptors C5AR, C3AR and complement components C1QA, C1QB, and C1QC are actively transcribed in G1 (). In contrast, examination of transcripts upregulated in S/G2/M reveal signaling pathways regulating the G2/M DNA damage checkpoint, mitotic roles polo-like kinases and melatonin degradation (). Subsequently, we observe ESPL1, PLK1, CDK1 and TOP2A transcripts associated with the S/G2/M phases of the cell cycle (). We confirmed the complement component expression via QPCR (). Overall, when comparing between age groups, we find expression of complement genes in E11.5 G1 IAHC cells suggesting this may be a critical pathway for the maturation of IAHC cells toward definitive HSCs resulting in adult engraftment, chemotactic and migration programs.
Discussion
We set out to determine how IAHCs are formed following their emergence from the endothelium. Our data reveal that the cell cycle length of E10.5 IAHC cells is approximately 5 hours. In addition, our clonal labeling analysis suggests that more than one hemogenic clone, (likely 2) in the dorsal aortic floorCitation33 contributes to the formation of a single IAHC. Several IAHCs may then be produced from multiple clones, as clonal labeling in the zebrafish suggests the existence of up to 30 HSC clones per aorta.Citation34 Clonal output is also likely heterogenous, as recent work using limited dilutional analyses suggests, with increased heterogeneous HSPC populations at E10 vs. E11.Citation42 Thus, IAHC formation is likely driven by the rapid cell proliferation of several hemogenic endothelial clones with differing functional capacities. These findings further support a recent observation that an initial pool of pre-HSCs is established, from which HSCs mature from by E11.5.Citation43 Correspondingly, the length of the cell cycle in E11.5 IAHC cells increases to about 8 hours. This observation is intriguing as fetal liver (FL) HSCs have been observed to have a mean generation time of 10.6 hours.Citation36
The cell cycle of FL and bone marrow (BM) HSCs is tightly associated with their ability to self-renew and differentiate.Citation44 The progression of HSCs through the cell cycle both in vitro and in vivo is accompanied by notable changes in their engraftment potential.Citation36,39-41,45-47 Several lines of evidence suggest that cell cycle position may influence repopulation activity.Citation36,39-41,45-47 Specifically, FL and BM HSCs in the G0/G1 appear to engraft adult recipients better than their S/G2/M counterparts.Citation36,39 Moreover, a permissive environment is also required for successful engraftment. Arora and colleagues demonstrated that embryonic (AGM) HSCs engraft neonatal recipients better than adult recipients.Citation37 They also found that adult-like (BM and FL) HSCs more efficiently reconstitute adult recipients than neonates.Citation37 Here, we investigated whether embryonic HSCs from ontogeny are at a specific cell cycle phase which may affect their engraftment in the adult BM. We note that adjusting for cell cycle phase at E10.5 (by selection and transplantation of the minority G1 population) does not overcome the inability of these cells to engraft adult hosts. However, the lengthening of cell cycle from E10.5 to E11.5 may be commiserate with the E11.5 IAHC cell repopulation ability. Our data show that E11.5 G1 IAHC populations may engraft better than their S/G2/M counterpart. This finding is in agreement with a recent report from Batsivari and colleagues who found that direct transplantation of mature E11.5 dHSCs at G0/G1 engrafted better than those in S/G2/M.Citation48 Furthermore, the seemingly coincident ability of E11.5 IAHC cells to engraft adult irradiated recipients point to the possibility that the lengthening of the cell cycle may influence HSC maturation. The number of IAHC cells detected during the hemogenic window is small with only ∼1500 – 2000 cells in an E10.5 AGM.Citation19 In our hands, the average percentage of cells that exhibit either FUCCI-Green or FUCCI-Red fluorescence ranges from 600 to 700 at E10.5, and 800 to 1000 cells at E11.5 per AGM. Thus, a significant limitation of our transplantation study is that only a limited number of cells could be harvested from any single litter for transplantation. We were able to transplant as low as ∼200 cells and as much ∼2000 cells per adult mouse (∼1/8 to 1 IAHC embryo equivalent EE) under these conditions, and thus may be at the detection boundary of the assay.
RNA sequencing of IAHC cells at E10.5 and E11.5 at different phases of the cell cycle revealed a dynamic transcriptional landscape that changes with developmental age and cell cycle phase. Across developmental age by E11.5, an enrichment for genes that regulate “cell movement,” “migration” and “inflammatory response” become evident and further support the findings Mckinney-Freeman and colleagues that IAHC cells may resemble a “macrophage-like signature”Citation49; in addition to the recent wealth of information pointing to inflammatory signals being critical for early HSC development.Citation50-52 Furthermore, since it has been hypothesized that IAHC cells migrate to the fetal liver after their formation in the AGM, it is likely that the maturation process of E10.5 IAHC cells may involve the activation of cell movement and cell migration signaling pathways via inflammatory signaling that prepares IAHCs for entry into circulation.
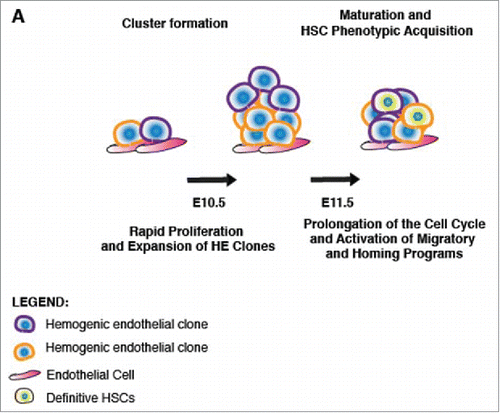
Transplantation of E11.5 G1 vs. S/G2/M IAHC cell populations suggested an advantage of G1 IAHC cells to engraft irradiated adult recipients, albeit with low numbers. This observation is in line with earlier findings that HSCs in G1 have an engraftment advantage over their S/G2/M counterparts.Citation36,39,40 Our findings also differed from a recent report that found that pre-HSCs in S/G2/M were capable of enhanced engraftment than pre-HSCs in G0/G1.Citation53 This discrepancy with recent work may be due to co-culturing which may alter cell cycle states. Transcriptional profiling of E11.5 G1 IAHC cells revealed an enrichment for transcripts that regulated the “immune response,” “chemotaxis” and “cell migration.” While not surprising, profiling of S/G2/M E11.5 IAHC cells demonstrated an activation of signaling pathways that regulated “cell growth” and “proliferation.” Thus, our data suggests that during the G1 phase of the cell cycle, IAHC cells may become sensitive to a multitude of cellular cues which include immune and inflammatory signals that may regulate cell movement, egress into circulation, and consequently, migration toward the fetal liver. Furthermore, there is evidence to suggest complement activation plays a role in donor HSPC migration and homing post transplantation.Citation54-56 It has been shown that C1q enhances the responsiveness of HSPCs to the chemoattractant CXCL−12 (SDF-1).Citation57 We find that genes of the classical complement cascade which include C1QA, C1QB and C1QC, as well as anaphylatoxin receptors C3AR and C5AR, to be upregulated in E11.5 G1 IAHC cells. Thus, the increased expression of complement system components in E11.5 G1 IAHC cells may provide a method for homing to the adult bone marrow, and possibly explain the engraftment potential of G1 IAHC cells relative to their S/G2/M counterparts. Overall, our data support a model following EHT where at E10.5, IAHCs form rapidly from the proliferation of at least 2 hemogenic clones, and subsequently cells comprising IAHCs undergo prolongation of their cell cycle length and activate transcriptional programs of migration and homing, which may lead to facilitation of transit into the fetal liver (). These findings further lend credence to the establishment of a robust HSC program following EHT, such that the very properties (prolonged cell cycle length, engraftment in G0/G1, lengthening of G1) that define fetal liver and adult HSCs is reflected in the newly emerged embryonic AGM HSCs.
Figure 7. Model of cluster formation and maturation of IAHCs to HSCs. (A) A scheme depicting a model of IAHC formation and maturation. During the hemogenic window (E10-E11), distinct hemogenic clones are present in the dorsal aorta which undergo rapid proliferation forming polyclonal IAHCs. Following formation of an IAHC, cells undergo maturation toward an HSPC phenotype which may include prolongation of the cell cycle and activation of migratory and homing programs.
Materials and methods
Mice
Mice were housed under a barrier facility following IACUC procedures and animal protocols under the University of California San Francisco Laboratory Animal Rersearch Committee Guidelines. C57/Bl6 (Wildtype), Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (R26r-mTmG),Citation58 Tg(Cdh5-cre)1Spe/J (VEC-Cre),Citation5 and Gt(ROSA)26Sortm1(CAG Brainbow2.1)Cle/J(R26r-confetti)Citation59 mice were obtained from Jackson Laboratories. Cdh5(PAC)-CreERT2 (Tg(Cdh5-cre/ERT2)1Rha)Citation60 mouse line was obtained from Ralph Adams, PhD, CancerUK, and Fucci Green Tg(CAG mAG/GMNN)504Amiy and Fucci Red Tg(CAG-mKO2/CDT1)596Amiy lines from RIKEN35. Males from this line were crossed to female homozygous Rosa26-mT/mG females. Pregnancies were timed in accordance with the vaginal plug (gestation day 0.5 on the day of the plug). Genomic DNA from adult ear punches, or conceptus yolk sacs was genotyped using MyTaq Extract PCR Kit (Bioline, BIO21127).
Low density clonal labeling
Low density clonal labeling was determined by titrating doses of tamoxifen beginning with 50 µg, 100 µg to 150µg. Tamoxifen was administered at E8.5 before any IAHC formation. After determining the tamoxifen dose, females were injected intraperitoneally with 0.1 mg at E8.5 before cluster formation. Embryos were evaluated at E10.5 (35–39sp) and E11.5 (44–48sp).
Tamoxifen preparation
Tamoxifen was purchased from MP Biologicals LLP. A 10 mg/ml stock was prepared by dissolving 10 mg of tamoxifen in 100 µl of 100% ethanol and mixed with 900 μl sterile sunflower oil (1:9). For low dose clonal labeling experiments, a stock solution of 2.5 mg/ml of tamoxifen was prepared in similar fashion.
Cell cycle length determination
Cell cycle length was determined by sequential administration of EdU and BrdU. First, 0.15 ml of EdU (10 mg/ml) was administered intraperitoneally. After 1.5 hours, 0.20 ml of BrdU (10 mg/ml) was injected. BrdU was detected by treating tissue sections with 2N HCl for 30 minutes at 37°C and incubated with 1:100 anti-BrdU (MOBU-1, Life Technologies #B35128). Samples were washed 3x with 1xPBS + 0.05% Tween (PBST) and a secondary antibody was applied at 1:200 (Invitrogen, #A21203). EdU was detected using a Click-iT EdU Detection System (Life Technologies, #C10337). This technique allows for identification of cells that are in S-phase (S) by EdU+ incorporation. Incorporation of both EdU+ and BrdU+ designates cells in the S phase (S). Incorporation of EdU+, and not subsequently BrDU−, delineates cells that are leaving the S-phase (L) during the 1.5 hrs between EdU and BrdU pulses. All proliferating IAHC cells are marked by PU.1, (P). The total length of S phase (Ts) is estimated by first calculating the number of cells leaving the S-phase (L) over the total number of cells in the S-phase (S). The initial time (Ti) between EdU and BrdU (1.5 hrs) is then divided by this fraction, thus Ts = Ti/(L/S). The total length of the cell cycle (Tc) is estimated by first determining the number of cells in the S-phase (S) over the total number of proliferating cells (P). The total S-phase length (Ts) is then divided by this fraction, thus Tc = Ts/(S/P).
Flow cytometry
Whole embryos or AGMs were dissociated as described previously (Lizama et al. 2014) and stained for 30 min at 4°C with agitation. Single cell suspensions were sorted in a BD FACS Aria III. Flow cytometric analyses were performed on a FACS Verse or FACS Aria III with FACSDiva 8.0 software (BD Biosciences) and data analyzed using FlowJo v10.0.7 (Tree Star). Antibodies were purchased from BD Biosciences: CD117-APC (#553356), CD31-Percp Cy5.5 (#562861), V450-SSEA-1 (#561561), APC-CD45.2 (#558702), FITC-CD45.1 (#553775), PeCy7-CD11b (#552850), FITC-CD3 (#55274), PE-Ter119 (#553673). Isotype controls used were: IgG2b-APC (#55391), IgM-V450 (#560861), IgG1-PercpCy5.5 (#562861), IgG2b-PeCy7 (#552849), IgG2b-FITC (#553988), IgG2b-PE (#553989). Antibodies were purchased from Biolegend: PE-CD45R/B220 (#103208) and IgG2a-PE (#400507).
Immunofluorescence and confocal microscopy
E10.5 – E11.5 embryos were fixed in 4% paraformaldehyde (PFA) solution at room temperature for 20 minutes or in 2% PFA overnight. Samples were frozen in Tissue-Tek OCT Compound (Sakura Finetek, 4583). 20–30 µm cryosections were obtained (Thermo Scientific Micron, HM550). Slides were incubated in blocking buffer (PBS, 0.5% Triton-X100) with 5% serum for 1 hr. Primary antibodies were incubated at 4°C overnight or room temperature for 2 hrs in blocking buffer. Slides were washed (PBS 0.05% Tween), incubated with the secondary antibodies for 1 hr, stained with 2μg/ul DAPI and mounted in Vectamount (Vector Laboratories, H-5000). Images were captured on a Leica SPE Confocal Microscope compiled using Imaris 8.6 (Bitplane; Belfast, UK) software.
Image analysis and cluster quantification
Immunofluorescence of R26R-mTmG and wild type sections were taken on a Leica SPE confocal microscope with a 40x oil objective. Immunofluorescence of FUCCI sections were taken on Leica SP5 confocal microscope with a 40x oil objective. All reconstructions were generated from z-stacks using the Imaris software [Imaris 8.6] (Bitplane; Belfast, UK). Images are a maximum projection of 30–40μm cryoslices. A Gaussian filter is applied to all images. IAHC cluster cell number counts were done based on nuclear PU.1, with endothelial labeling of Sox17 with the addition of CD31.
Immunofluorescence antibodies
For immunofluorescence experiments 1:100 of Rabbit Runx1 (ab35962), Goat Sox17 (R&D Systems #AF1924) Rat CD117 Clone (2B8) BD PharMingen (#553352) Rat CD31 Clone (Mec13.3) BD PharMingen (#553370) Rabbit PU.1 Clone (9G7) Cell Signaling (#2258) were used. Secondary antibodies according to host species were used at 1:200 dilutions (Invitrogen: donkey anti-goat 488 #A11055, donkey anti-goat #A21447, donkey anti-rat 594 #A21209, donkey anti-rabbit 647 #A31573, Abcam: donkey anti-rabbit 405 #175649).
RNA isolation and sequencing
Samples were pooled from 2–3 litters. Total RNA was isolated and purified from sorted cells using the QIAGEN RNeasy Plus Micro Kit (#74034) following manufacturer's instructions. RNA integrity was determined using a Bioanalyzer. Samples were ribo-depleted and amplified with Ovation RNA-Seq System V2. Paired-end 2 × 100 bp sequencing was performed using the Illumina HiSeq2000 machine. RNA sequencing analysis was done using Galaxy (https://usegalaxy.org/u/laiumiunix/w/rnaseqjoandata) with a False discovery rate (FDR) of 0.085. Transcripts with ≥ 2-fold change expression were further analyzed using the Ingenuity Pathway Analysis to identify key biologic functions and pathways that are expected to be more active in E11.5 than E10.5. To be more stringent, we focused our analysis to functions and pathways with a positive z-score. Data available under GEO accession number: GSE 99314.
Transplantation
Eight–12 week old CD45.2 C57Bl/6 FUCCI mice were used as donors for cell isolation. E10-E10.5 (30–39sp) and E11-E11.5 (40–48sp) AGMs were sorted for SSEA-1−/CD31+/CD117+ FUCCI-Red or FUCCI-Green as donor cells. 8–12 week old CD45.1 C57Bl/6-Boy/J wild type mice were used as recipients for cell transplantation. Congenic recipient mice (CD45.1) were irradiated with 900 rads, split dose, 3 hours apart using a cesium source. Purified donor cells were injected intravenously with 200,000 spleen helper cells, and haematopoietic reconstitution was monitored over time in the peripheral blood based on CD45.2 expression. Recipients with ≥1% donor chimerism were considered reconstituted. Transplanted mice were kept on antibiotic-containing food for 2 weeks. Recipients of helper cells and congenic donor cells were used as negative control, and recipients of 4 × 106 CD45.2 whole bone marrow cells were used as positive control. All mice were maintained at UCSF in accordance with IACUC approved protocols.
QPCR
Total RNA was isolated from pooled littermates of wild-type AGM SSEA-1−/CD31+/CD117+ populations (∼2000 cells) from E10.5 and E11.5 as described previously. A total of 40 µg of RNA was transformed into CDNA using SuperScript VILO Master Mix (Thermo Fisher Scientific #11755–050) following the manufacturer's instructions.
Statistics
All the data are expressed as mean ± SD. P values were generated either with a Fisher's exact test or an unpaired t-test with Welch's correction and considered significant when ≤ 0.05. “n” indicates the numbers of independent experiments performed. Clonality data from the low dose fate tracing experiments were binned according to IAHC size Type A (3–9 cells) and Type B (>10 cells), and fisher's exact test was performed to evaluate the correlation between IAHC size and clonality.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
JPZ and ACZ designed and planned experiments. JPZ performed the experiments, with assistance from COL and KMCR. JPZ and ACZ wrote the manuscript.
KCCY_S_1361569.zip
Download Zip (451.6 KB)Acknowledgments
We would like to acknowledge the UCSF Core Laboratory for Cell Analysis supported by the National Cancer Institute Cancer Center Support Grant (5P30CA082103), and Ajay Chawla M.D., Ph.D. for use of his laboratory flow cytometer. We also thank Atsushi Miyawaki, Ph.D. of the RIKEN institute, and Ralph Adams, PhD, of CancerUK, for generously sharing the transgenic mouse lines. We also would like to acknowledge Hui Yu, Ph.D. for his help in animal husbandry and genotyping.
Funding
This work was supported by the NSF and CIRM pre-doctoral fellowships (JPZ), the Burroughs Wellcome Fund CAMS award (ACZ, 1008408.01), and the NIH Innovator Award program (ACZ, 1DP2HL117743–01).
Related Research Data
References
- Jaffredo T, Gautier R, Eichmann A, Dieterlen-Lièvre F. Intraaortic hemopoietic cells are derived from endothelial cells during ontogeny. Development. 1998;125(22):4575-83. PMID:9778515
- Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86(6):897-906. PMID:8808625
- Zovein AC, Hofmann JJ, Lynch M, French WJ, Turlo KA, Yang Y, Becker MS, Zanetta L, Dejana E, Gasson JC, et al. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3(6):625-36. doi:10.1016/j.stem.2008.09.018. PMID:19041779
- North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marín-Padilla M, Speck NA. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126(11):2563-75. PMID:10226014
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457(7231):887-91. doi:10.1038/nature07619. PMID:19129762
- Boisset J-C, Andrieu-Soler C, van Cappellen WA, Clapes T, Robin C. Ex vivo time-lapse confocal imaging of the mouse embryo aorta. Nat Protoc. 2011;6(11):1792-805. doi:10.1038/nprot.2011.401. PMID:22036882
- de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 2000;19(11):2465-74. doi:10.1093/emboj/19.11.2465. PMID:10835345
- North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, Binder M, Dzierzak E, Speck NA. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16(5):661-72. PMID:12049718
- de Bruijn MFTR, Ma X, Robin C, Ottersbach K, Sanchez M-J, Dzierzak E. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity. 2002;16(5):673-83. PMID:12049719
- Garcia-Porrero JA, Godin IE, Dieterlen-Lièvre F. Potential intraembryonic hemogenic sites at pre-liver stages in the mouse. Anat Embryol. 1995;192(5):425-35. PMID:8546334
- Ottersbach K, Dzierzak E. The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev Cell. 2005;8(3):377-87. doi:10.1016/j.devcel.2005.02.001. PMID:15737933
- Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HKA. The placenta is a niche for hematopoietic stem cells. Dev Cell. 2005;8(3):365-75. doi:10.1016/j.devcel.2004.12.016. PMID:15737932
- Padrón-Barthe L, Temiño S, Villa del Campo C, Carramolino L, Isern J, Torres M. Clonal analysis identifies hemogenic endothelium as the source of the blood-endothelial common lineage in the mouse embryo. Blood. 2014;124(16):2523-32. doi:10.1182/blood-2013-12-545939. PMID:25139355
- Yzaguirre AD, Speck NA. Insights into blood cell formation from hemogenic endothelium in lesser-known anatomic sites. Dev Dyn. 2016;245(10):1011-28. doi:10.1002/dvdy.24430. PMID:27389484
- Lux CT, Yoshimoto M, McGrath K, Conway SJ, Palis J, Yoder MC. All primitive and definitive hematopoietic progenitor cells emerging before E10 in the mouse embryo are products of the yolk sac. Blood. 2008;111(7):3435-38. doi:10.1182/blood-2007-08-107086. PMID:17932251
- McGrath KE, Frame JM, Fegan KH, Bowen JR, Conway SJ, Catherman SC, Kingsley PD, Koniski AD, Palis J. Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep. 2015;11(12):1892-904. doi:10.1016/j.celrep.2015.05.036. PMID:26095363
- de Bruijn MF, Peeters MC, Luteijn T, Visser P, Speck NA, Dzierzak E. CFU-S(11) activity does not localize solely with the aorta in the aorta-gonad-mesonephros region. Blood. 2000;96(8):2902-04. PMID:11023528
- Gordon-Keylock S, Sobiesiak M, Rybtsov S, Moore K, Medvinsky A. Mouse extraembryonic arterial vessels harbor precursors capable of maturing into definitive HSCs. Blood. 2013;122(14):2338-45. doi:10.1182/blood-2012-12-470971. PMID:23863896
- Yokomizo T, Dzierzak E. Three-dimensional cartography of hematopoietic clusters in the vasculature of whole mouse embryos. Development. 2010;137(21):3651-61. doi:10.1242/dev.051094. PMID:20876651
- Boisset JC, Clapes T, Klaus A, Papazian N, Onderwater J, Mommaas-Kienhuis M, Cupedo T, Robin C. Progressive maturation toward hematopoietic stem cells in the mouse embryo aorta. Blood. 2015;125(3):465-69. doi:10.1182/blood-2014-07-588954. PMID:25301706
- Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464(7285):112-15. doi:10.1038/nature08761. PMID:20154732
- Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464(7285):108-11. doi:10.1038/nature08738. PMID:20154733
- Taoudi S, Gonneau C, Moore K, Sheridan JM, Blackburn CC, Taylor E, Medvinsky A. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE-cadherin+CD45+ pre-definitive HSCs. Cell Stem Cell. 2008;3(1):99-108. doi:10.1016/j.stem.2008.06.004. PMID:18593562
- Müller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. 1994;1(4):291-301. PMID:7889417
- Taylor E, Taoudi S, Medvinsky A. Hematopoietic stem cell activity in the aorta-gonad-mesonephros region enhances after mid-day 11 of mouse development. Int J Dev Biol. 2010;54(6–7):1055-60. doi:10.1387/ijdb.103152et. PMID:20711982
- Rybtsov S, Batsivari A, Bilotkach K, Paruzina D, Senserrich J, Nerushev O, Medvinsky A. Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(-) embryonic precursor. Stem Cell Reports. 2014;3(3):489-501. doi:10.1016/j.stemcr.2014.07.009. PMID:25241746
- Wilkinson AC, Kawata VK, Schütte J, Gao X, Antoniou S, Baumann C, Woodhouse S, Hannah R, Tanaka Y, Swiers G, et al. Single-cell analyses of regulatory network perturbations using enhancer-targeting TALEs suggest novel roles for PU.1 during haematopoietic specification. Development. 2014;141(20):4018-30. doi:10.1242/dev.115709. PMID:25252941
- Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. Detection of S-phase cell cycle progression using 5-ethynyl-2“-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2-”deoxyuridine antibodies. BioTechniques. 2008;44(7):927-29. doi:10.2144/000112812. PMID:18533904
- Cappella P, Gasparri F, Pulici M, Moll J. A novel method based on click chemistry, which overcomes limitations of cell cycle analysis by classical determination of BrdU incorporation, allowing multiplex antibody staining. Cytometry A. 2008;73(7):626-36. doi:10.1002/cyto.a.20582. PMID:18521918
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311-22. doi:10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. PMID:10653597
- Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol. 2005;283(1):113-27. doi:10.1016/j.ydbio.2005.04.005. PMID:15893304
- Nomura T, Gotoh H, Ono K. Changes in the regulation of cortical neurogenesis contribute to encephalization during amniote brain evolution. Nat Commun. 2013;4:2206. doi:10.1038/ncomms3206. PMID:23884180
- Chen MJ, Li Y, De Obaldia ME, Yang Q, Yzaguirre AD, Yamada-Inagawa T, Vink CS, Bhandoola A, Dzierzak E, Speck NA. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. 2011;9(6):541-52. doi:10.1016/j.stem.2011.10.003. PMID:22136929
- Henninger J, Santoso B, Hans S, Durand E, Moore J, Mosimann C, Brand M, Traver D, Zon L. Clonal fate mapping quantifies the number of haematopoietic stem cells that arise during development. Nat Cell Biol. 2017;19(1):17-27. doi:10.1038/ncb3444. PMID:27870830
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487-98. doi:10.1016/j.cell.2007.12.033. PMID:18267078
- Nygren JM, Bryder D, Jacobsen SEW. Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J Immunol. 2006;177(1):201-08. PMID:16785515
- Arora N, Wenzel PL, McKinney-Freeman SL, Ross SJ, Kim PG, Chou SS, Yoshimoto M, Yoder MC, Daley GQ. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev Cell. 2014;29(5):621-28. doi:10.1016/j.devcel.2014.04.013. PMID:24914562
- Taoudi S, Medvinsky A. Functional identification of the hematopoietic stem cell niche in the ventral domain of the embryonic dorsal aorta. Proc Natl Acad Sci USA. 2007;104(22):9399-03. doi:10.1073/pnas.0700984104. PMID:17517650
- Fleming WH, Alpern EJ, Uchida N, Ikuta K, Spangrude GJ, Weissman IL. Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J Cell Biol. 1993;122(4):897-902. PMID:8349737
- Orschell-Traycoff CM, Hiatt K, Dagher RN, Rice S, Yoder MC, Srour EF. Homing and engraftment potential of Sca-1(+)lin(-) cells fractionated on the basis of adhesion molecule expression and position in cell cycle. Blood. 2000;96(4):1380-87. PMID:10942381
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96(6):3120-25. PMID:10077647
- Ganuza M, Hadland B, Chabot A, Li C, Kang G, Bernstein I, McKinney-Freeman S. Murine hemogenic endothelial precursors display heterogeneous hematopoietic potential ex vivo. Exp Hematol. 2017;51:25-35.e6. doi:10.1016/j.exphem.2017.04.006. PMID:28450163
- Rybtsov S, Ivanovs A, Zhao S, Medvinsky A. Concealed expansion of immature precursors underpins acute burst of adult HSC activity in foetal liver. Development. 2016;143(8):1284-89. doi:10.1242/dev.131193. PMID:27095492
- Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709-20. doi:10.1083/jcb.201102131. PMID:22123859
- Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116(10):2808-16. doi:10.1172/JCI28310. PMID:17016561
- Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0). Blood. 2000;96(13):4185-93. PMID:11110690
- Habibian HK, Peters SO, Hsieh CC, Wuu J, Vergilis K, Grimaldi CI, Reilly J, Carlson JE, Frimberger AE, Stewart FM, et al. The fluctuating phenotype of the lymphohematopoietic stem cell with cell cycle transit. J Exp Med. 1998;188(2):393-98. PMID:9670051
- Batsivari A, Rybtsov S, Souilhol C, Binagui-Casas A, Hills D, Zhao S, Travers P, Medvinsky A. Understanding hematopoietic stem cell development through functional correlation of their proliferative status with the intra-aortic cluster architecture. Stem Cell Reports. 2017;8(6):1549-62. doi:10.1016/j.stemcr.2017.04.003. PMID:28479304
- McKinney-Freeman S, Cahan P, Li H, Lacadie SA, Huang HT, Curran M, Loewer S, Naveiras O, Kathrein KL, Konantz M, et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell. 2012;11(5):701-14. doi:10.1016/j.stem.2012.07.018. PMID:23122293
- Sawamiphak S, Kontarakis Z, Stainier DYR. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell. 2014;31(5):640-53. doi:10.1016/j.devcel.2014.11.007. PMID:25490269
- Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM, Yzaguirre AD, Cai X, Cortes M, Maijenburg MW, et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014;28(23):2597-612. doi:10.1101/gad.253302.114. PMID:25395663
- Espín-Palazón R, Stachura DL, Campbell CA, García-Moreno D, Del Cid N, Kim AD, Candel S, Meseguer J, Mulero V, Traver D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014;159(5):1070-85. doi:10.1016/j.cell.2014.10.031. PMID:25416946
- Zhou F, Li X, Wang W, Zhu P, Zhou J, He W, Ding M, Xiong F, Zheng X, Li Z, et al. Tracing haematopoietic stem cell formation at single-cell resolution. Nature. 2016;533(7604):487-92. doi:10.1038/nature17997. PMID:27225119
- Reca R, Mastellos D, Majka M, Marquez L, Ratajczak J, Franchini S, Glodek A, Honczarenko M, Spruce LA, Janowska-Wieczorek A, et al. Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing-related responses to SDF-1. Blood. 2003;101(10):3784-93. doi:10.1182/blood-2002-10-3233. PMID:12511407
- Ratajczak J, Reca R, Kucia M, Majka M, Allendorf DJ, Baran JT, Janowska-Wieczorek A, Wetsel RA, Ross GD, Ratajczak MZ. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood. 2004;103(6):2071-78. doi:10.1182/blood-2003-06-2099. PMID:14604969
- Lee HM, Wu W, Wysoczynski M, Liu R, Zuba-Surma EK, Kucia M, Ratajczak J, Ratajczak MZ. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia. 2009;23(11):2052-62. doi:10.1038/leu.2009.158. PMID:19657368
- Jalili A, Marquez-Curtis L, Shirvaikar N, Wysoczynski M, Ratajczak M, Janowska-Wieczorek A. Complement C1q enhances homing-related responses of hematopoietic stem/progenitor cells. Transfusion. 2010;50(9):2002-10. doi:10.1111/j.1537-2995.2010.02664.x. PMID:20456695
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593-605. doi:10.1002/dvg.20335. PMID:17868096
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450(7166):56-62. doi:10.1038/nature06293. PMID:17972876
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465(7297):483-86. doi:10.1038/nature09002. PMID:20445537