
ABSTRACT
Cell cycle reactivation in adult neurons is an early hallmark of neurodegeneration. The lipopolysaccharide (LPS) is a well-known pro-inflammatory factor that provokes neuronal cell death via glial cells activation. The retinoblastoma (RB) family includes RB1/p105, retinoblastoma-like 1 (RBL1/p107), and retinoblastoma-like 2 (Rb2/p130). Several studies have indicated that RB proteins exhibit tumor suppressor activities, and play a central role in cell cycle regulation. In this study, we assessed LPS-mediated inflammatory effect on cell cycle reactivation and apoptosis of neuronally differentiated cells. Also, we investigated whether the LPS-mediated inflammatory response can influence the function and expression of RB proteins. Our results showed that LPS challenges triggered cell cycle reactivation of differentiated neuronal cells, indicated by an accumulation of cells in S and G2/M phase. Furthermore, we found that LPS treatment also induced apoptotic death of neurons. Interestingly, we observed that LPS-mediated inflammatory effect on cell cycle re-entry and apoptosis was concomitant with the aberrant expression of RBL1/p107 and RB1/p105. To the best of our knowledge, our study is the first to indicate a role of LPS in inducing cell cycle re-entry and/or apoptosis of differentiated neuronal cells, perhaps through mechanisms altering the expression of specific members of RB family proteins. This study provides novel information on the biology of post-mitotic neurons and could help in identifying novel therapeutic targets to prevent de novo cell cycle reactivation and/or apoptosis of neurons undergoing neurodegenerative processes.
Introduction
Central nervous system (CNS) is formed by diverse populations of neurons, which are generated from a pool of multipotent precursors during the embryogenesis.Citation1 The generation of post-mitotic neuronal cells requires mechanisms that temporally coordinate both the cell cycle exit and the differentiation program.Citation2 When multipotent cells withdraw from the cell cycle, they enter the post-mitotic G0, losing their proliferative capability.Citation3
Although mature neurons are considered non-proliferative cells, aberrant cell cycle re-entry has been demonstrated in different neuronal populations, including sympathetic and cortical neurons, when subjected to acute insults such as neurotrophic factor deprivation, oxidative stress, and excitotoxicity among other.Citation4,Citation5
Cell cycle reactivation in adult neurons is an early hallmark of neurodegenerationCitation6 and brain injury.Citation7-9 Evidence of this aberrant re-entry into the cell cycle in the neurodegeneration includes the abnormal re-expression of various cyclins, CDKs and CDKIs that potentially lead to the active expression of growth-promoting genes.Citation5 However, the mechanism that drives post-mitotic neurons to re-enter cell cycle remains elusive.
Inflammatory response contributes to the pathogenesis of neurodegenerative disorders.Citation10,Citation11 The lipopolysaccharide (LPS) is a well-known pro-inflammatory factor that sensitizes the brain to hypoxia injury.Citation12,Citation13 It has been reported, that LPS elicits neuronal cell death via glial cells activation.Citation14-16 However, evidence of a direct involvement of LPS in neuronal apoptosis is lacking.
The retinoblastoma (RB) family includes RB1/p105, retinoblastoma-like 1 (RBL1/p107), and retinoblastoma-like 2 (Rb2/p130). Several studies have indicated that RB proteins exhibit tumor suppressor activities, and play a central role in cell cycle regulation.Citation17
The first in vivo evidence that RB proteins are involved in maintaining the survival of postmitotic neurons arise from observations that disruption of the RB1/p105 causes profound defects in neurogenesis of mouse embryos.Citation18-21
Successive studies have shown that Rb2/p130 associated with the E2F4 transcription factor in neurons, and Rb2/p130–E2F4 complex recruits the chromatin modifiers HDAC1 and Suv39H1 to promote gene silencing and neuron survival.Citation22 Furthermore, apoptotic stimuli induce neuron death by causing Rb2/p130 hyperphosphorylation, and successive disruption of Rb2/p130–E2F4 – HDAC1-Suv39H1 complexes.Citation22
Furthermore, investigations have indicated that RBL1/p107 promotes the differentiation program of neural progenitor toward a neuronal fate.Citation23,Citation24
RB proteins are not simple oncosuppressor, but they exhibit a pleiotropic function in different biologic systems,Citation25-27 including the Central Nervous System (CNS).Citation21 Here, we demonstrated that LPS – induced inflammatory response triggers a disabled G1/S checkpoint, cell cycle reactivation, and apoptosis in murine neuronal cells. Moreover, LPS challenge provokes an aberrant RB proteins expression in these cells.
Results
Effect of LPS treatment on cell morphology
Phase-contrast microscopy revealed that acute LPS treatment induced changes in the average of neurite length and number with respect to unstimulated NE-4C-RA cells (, ). Importantly, chronic LPS treatment significantly decreased the number of branching points per cell ().
Figure 1. Effect of LPS treatment on cell morphology. Upper panels (A, B, and C) show phase-contrast microscopy analysis of treated and untreated neuronal differentiated NE-4C cells. Differentiation was achieved by treating NE-4C cells with 100 nM RA for 48h in all conditions (NE-4C-RA). Micrographs illustrate unstimulated cells (A) acute LPS-stimulated NE-4C-RA cells (B) and chronic LPS-stimulated NE-4C-RA cells (C). Middle (D, E, and F) and lower panels (G, H, and I) show the immunolocalization of the neuronal marker NF-H (Heavy Neurofilament) and the astrocyte marker GFAP (Glial Fibrillary Acidic Protein) respectively, in treated (E, F, H, and I) and untreated NE-4C-RA cells (D and G). Micrographs show unstimulated (D and G), acute LPS-stimulated (E and H) and chronic LPS-stimulated (F and I). LPS treatment significantly decreases neurite number and length NE-4C-RA cells. Data presented are representative of 3 independent experiments; (Bar 100µm).
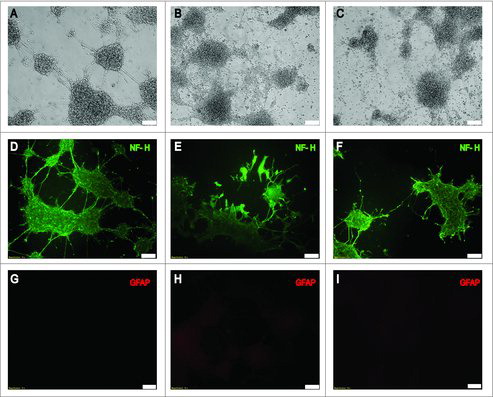
The purity of the cultures was assessed by indirect immunofluorescence for the neuronal marker Heavy Neurofilament (NF-H) () and the astroglial marker Glial Fibrillary Acidic Protein (GFAP) ().
An average of 98% of the cells was positive for the expression of NF-H and negative for GFAP in all the culture conditions examined in this study.
LPS triggers cell cycle reactivation of terminally differentiated neurons
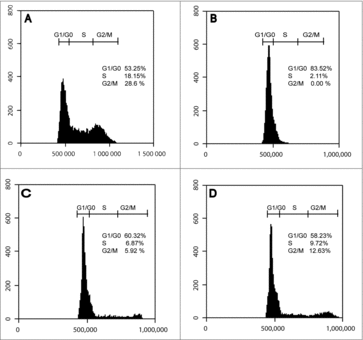
An increasing body of evidence has indicated that neurons under aberrant stimuli, including environmental factors, oxidative stress, inflammation, neurotrophic deprivation, and excitotoxicity, can actively re-enter the cell cycle, replicate DNA and survive as tetraploid, or die by apoptosis.Citation4,Citation5,Citation8 However, the mechanisms that drive post-mitotic neurons to re-enter cell cycle remain elusive, and such it is the role played by the tetraploid neurons.Citation8 We assessed the effect of acute or chronic LPS challenges on cell cycle reactivation. The cell cycle of cycling NE-4C, neuronal differentiated NE-4C-RA, and LPS-treated NE-4C-RA cells was analyzed using flow cytometry. As expected, the results revealed that cycling NE-4C cells were under proliferative conditions (G1/G0 53.25%, S 18.15%, G2/M 28.6%) () while neuronally differentiated NE-4C-RA cells were arrested in G0/G1 phase (G1/G0 83.52%, S 2.11%, G2/M 0.00%) (). However, acute LPS treatment induced an accumulation of NE-4C-RA cells in S and G2/M phase compared with unstimulated NE-4C-RA cells (G1/G0 60.32%, S 6.87%, G2/M 5.92%) (). Interestingly, chronic LPS exposure induces a significant accumulation of cells in G2 phase (G1/G0 58.23%, S 9.72%, G2/M 12.63%) ().
Figure 2. Effect of LPS on Cell Cycle. Cell cycle analysis by flow cytometry of proliferating NE-4C cells (A), RA-differentiated NE-4C cells, NE-4C-RA, (B), acute LPS-stimulated NE-4C-RA cells (C) and chronic LPS stimulated NE-4C-RA cells (D). Exposure to RA caused cell cycle withdrawal of neuronal cells, indicated by G0/G1 arrest (A vs. B). LPS treatment induced accumulation of NE-4C-RA cells in S and G2/M phase (C and D vs. B). The accumulation of NE-4C-RA cells in S and G2/M phase was particularly pronounced under LPS chronic stimulation. Data presented are representative of 3 independent experiments run in triplicate.
Apoptosis assay of NE-4C-RA cells under acute or chronic LPS exposure
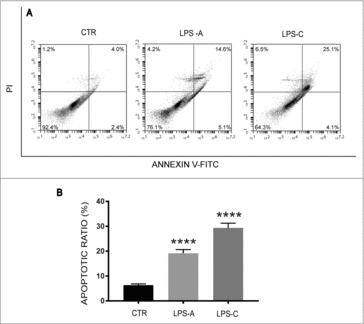
Cell cycle reactivation in adult neurons has been indicated as a hallmark of neurodegeneration as well as linked to apoptosis.Citation6,Citation28-30 We assessed whether LPS stimulation could induce apoptotic death of neurons besides to trigger cell cycle re-enter. Cell apoptosis was assessed using a standard flow cytometry-based Annexin V/PI apoptosis assay. Viable NE-4C-RA cells were both Annexin V and PI negative (92.4%) (); NE-4C-RA cells under acute LPS treatment showed reduced cell viability (76.1% vs. 92.4%) and a significant amount of cells in apoptosis (p = 0.0001) (, ). NE-4C-RA cells under chronic LPS treatment exhibited a more pronounced reduction of cell viability (64.3% vs. 92.4%) and a greater increase of cells in apoptosis (p = 0.0001) (, ), indicating an involvement of LPS in inducing apoptotic death of neurons.
Figure 3. Effect of LPS treatment on cell death. The figure shows apoptosis analysis in unstimulated neuronal differentiated NE-4C-RA cells (CTR), acute LPS-stimulated NE-4C-RA cells (LPS-A) and chronic LPS stimulated NE-4C-RA cells (LPS-C). Apoptosis was assessed using a standard flow cytometry-based Annexin V/PI apoptosis assay. As shown in , LPS treatment induced apoptotic cell death in both acute (LPS-A) and chronic condition (LPS-C). In , the ratio of apoptosis among different experimental groups was reported. Apoptosis ratio was early apoptosis percentage plus late apoptosis percentage. The data were presented as the means ± SEM. Columns, mean of three independent experiments; SEM; ****p ≤ 0.0001.
LPS treatment induces aberrant RB proteins expression
RB family proteins are known for their central role in regulating the cell cycle progression.Citation31,Citation32 Moreover, accumulating evidence suggests that RB proteins are involved in apoptosis, senescence, and differentiation.Citation17,Citation26,Citation33-35 Neurodegeneration has been linked to abnormal re-entry of neurons into the cell cycle.Citation6,Citation9,Citation36 However, the mechanism/s underlying this phenomenon and the role played by RB proteins in this process is still unclear.
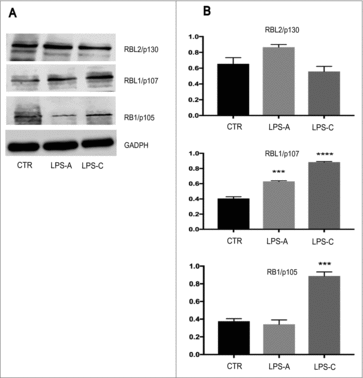
To assess the effects of acute or chronic LPS treatment on RB proteins expression, we performed Western blotting analysis by using total protein lysate from NE-4C-RA cells. Our results indicated that both acute and chronic LPS treatment induced a significant increase of RBL1/p107 protein level (LPS-A: p ≤ 0.0005; LPS-C: p ≤ 0.0001) ( and ). On the other hand, only chronic LPS treatment was able to induce a significant increase of RB1/p105 (LPS-C: p ≤ 0.0005), ( and ). Upon LPS treatment, no significant changes were observed in the expression of RBL2/p130 with respect to untreated NE-4C-RA cells ( and ).
Figure 4. Effect of LPS treatment of RB family protein expression. Western Blotting analysis was performed to assess the effect of acute LPS treatment (LPS-A) and chronic LPS treatment (LPS-C) on the expression of RBL2/p130, RBL1/p107, RB1/p105 in neuronal differentiated NE-4C-RA. As shown in and , both acute and chronic LPS treatment induced a significant increase in RBL1/p107 expression as compared to untreated NE-4C-RA cells (CTR). Only chronic LPS treatment was able to induce a significant increase in RB1/p105 expression. No significant effect was observed in RBL2/p130 expression after LPS treatment. The expression of GAPDH was assessed to normalize protein loading. The data were presented as the means ± SEM. Columns, mean of three independent experiments; bars, SEM; ***p ≤ 0.0005; ****p ≤ 0.0001.
Discussion
Unlike most cell types, neurons are typically considered post-mitotic cells that undergo apoptosis in response to cell cycle re-activation.Citation28 Interestingly, recent studies have suggested that cell cycle re-entry of neurons in the adult brain can be considered an early hallmark of neurodegeneration.Citation6,Citation8
Different mechanisms regulate G1/S and G2/M checkpoint in neuronal cells and are connected with the apoptotic machinery. There is evidence indicating that these mechanisms can be modulated by environmental signals, including neurotrophic deprivation, inflammation, oxidative stress and hypoxia, and these stimuli can induce either cell death or cell cycle reactivation of differentiated neurons.Citation4,Citation5
In this study, we assessed lipopolysaccharides (LPS)-mediated inflammatory effect on cell cycle reactivation and apoptosis of neuronal differentiated NE-4C cells. Moreover, we investigated whether the LPS-mediated inflammatory effect can influence the function and expression of RB family proteins. Our results showed that both acute and chronic LPS challenges caused cell cycle reactivation of differentiated neuronal NE-4C cells, indicated by an accumulation of NE-4C-RA cells in S and G2/M phase (). Furthermore, we found that LPS treatment also induced apoptotic death of neurons (). Interestingly, we observed that LPS-mediated inflammatory effect on cell cycle re-entry and apoptosis was concomitant with the aberrant expression of RB family proteins, in particular RBL1/p107 and RB1/p105 ().
In vitro and in vivo studies have indicated that administration of LPS causes brain microglia activation.Citation37,Citation38 Also, it has been reported that LPS challenge inhibits the proliferation of cultured neural stem/progenitor cellsCitation39 and that long-term LPS exposure causes an induction of nitric oxide (NO) in both NE-4C and differentiated NE-4C cells, thus eliciting an inflammatory response.Citation40
To the best of our knowledge, our study is the first to indicate a role of LPS in inducing cell cycle re-entry and/or apoptosis of differentiated neuronal cells, perhaps through mechanisms altering the expression of specific members of Rb family proteins.
Although, traditionally, the presence of Rb family proteins in adult neurons has been associated with a variety of differentiative functions (including neuronal maturation and migration, and synaptic plasticity), our study suggests that these proteins may play an essential role in the induction of cell cycle re-entry and/or apoptosis of mature neurons undergoing prolonged stress stimuli, such as LPS-mediated inflammatory condition. Further studies are required to deeply understand the complexity of the cross-talk among Rb family proteins, cell cycle regulation and apoptotic machinery in neurons enduring stress conditions. However, our study provides novel information with a high clinical significance. In fact, the knowledge gained in deciphering the role of RB proteins in brain inflammation has the potential to shed light on the mechanisms regulating the biology of post-mitotic neurons and could identify novel therapeutic targets to prevent de novo cell cycle reactivation and/or apoptosis of neurons undergoing neurodegenerative processes.
Materials and methods
Cell culture
Mouse neural stem cells (NE-4C) were purchased from ATCC (Manassas, VA). Cells were seeded in poly- L-lysine (15 ug/ml; Sigma-Aldrich, St. Louis, MO) coated plate and cultured in EMEM medium (ATCC) containing 10 % FBS (ATCC) and 4mM glutamine (ATCC) to maintain the proliferating condition.
Neuronal differentiation
Cell were seeded (1 × 104 cells cm2) in poly- L-lysine (100 ug/ml) coated plate and cultured in serum-free DMEM: F12 medium (ATCC) containing Insulin-transferrin-selenium supplement (Sigma-Aldrich). 24 h after seeding, cells were treated with Retinoic Acid (RA, f. c. 100 nM) for 48 h. RA was successively washed out, and cells were kept in culture with DMEM: F12 supplement for additional 4 days to obtain fully differentiated neurons (NE-4C-RA).
Treatments
NE-4C-RA cells were incubated with fresh medium containing bacterial endotoxin lipopolysaccharide (LPS) (Sigma-Aldrich) at 10 ng/ml (f. c.). LPS has been used to mimic neuronal inflammation in vitro. Acute and chronic stimulation was obtained following the protocol suggested by Cacci et al. 2008.Citation41 Briefly, untreated cells received fresh medium without LPS every 24 h over a 72 h period. Acutely stimulated NE-4C-RA cells received fresh medium without LPS for the first 48 h, and fresh medium containing LPS for the last 24 h of incubation. Chronically stimulated NE-4C-RA cells received a fresh medium containing LPS every 24 h.
Immunofluorescence
LPS-treated N4–2C-RA cells and untreated N4–2C-RA cells were grown on coverslips for 72 h. Immunofluorescence was performed according to the Immunofluorescence Application kit (Cell Signaling Technology, Danvers, MA). Cells were incubated with anti-rabbit Heavy Neurofilament (NF-H) (1:100; Cell Signaling) or anti-mouse GFAP (1:100; Cell Signaling), overnight at 4°C. The coverslips were rinsed with PBS and incubated with secondary antibodies for 1 h at room temperature. Secondary fluorochrome-conjugated antibodies were: goat anti-rabbit IgG AlexaFluor 488 and goat anti-mouse IgG AlexaFluor 555 (Thermo Fisher Scientific, Waltham, MA). The preparations were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and analyzed with an Olympus IX81 deconvolution fluorescence microscope (Olympus Microscopes, Tokyo, Japan). No staining was detected when conjugates alone were used as negative controls. Image magnification was 10x.
Western blotting
Protein extracts were resolved using SDS-PAGE and subsequently transferred onto nitrocellulose membrane (Sigma-Aldrich). For antibody detection, membranes were blocked in Phosphate Saline Buffer (PBS; Sigma-Aldrich) containing 5% fat-free milk powder (Sigma-Aldrich) for 20min at room temperature and then incubated with the following primary antibodies overnight: mouse anti-RBL2/p130 (BioScience, San Jose, CA), mouse-anti RBL1/p107 (Santa Cruz Biotechnology, Dallas, TX), mouse-anti RB1/p105 (Santa Cruz Biotechnology) and rabbit anti-GADPH (Santa Cruz Biotechnology). After washing, the membranes were incubated with the appropriate peroxidase-labeled secondary antibody (Santa Cruz Biotechnology) 1 h at room temperature and visualized using the Super Signal Western Dura substrate (Pierce, Rockford, USA)
Cell cycle analysis
LPS-treated N4–2C-RA cells (1 × 106/ml) and untreated N4–2C-RA cells (1 × 106/ml) were washed with PBS, fixed in 70% (v/v) ethanol and stored at 4°C, according to protocols well-established in the laboratory.Citation42 The cells were then centrifuged at 1,000 rpm, washed twice with PBS, and the pellets were resuspended in 0.4 ml of freshly prepared PBS solution containing 50 µg/ml of propidium iodide (PI) and 0.2 mg/ml of RNase (Abcam, Cambridge, UK)29. The stained samples were incubated 30 min at 37°C and analyzed by using an Accuri C6 Flow Cytometer (Accuri, Bioscience San Jose, CA). Performance validation of the instrument was performed following the manufacturer's instructions and with 6 and 8 peak fluorescent bead mixtures (Accuri). The analysis was based on light scatter and fluorescence signals produced from 20 mW laser illumination at 488 nm. Fluorescence signals were screened using an FL-2 filter, with a 585/40 nm band-pass filter. Statistical analysis was performed using Accuri C6 flow cytometer software. The flow cytometer was routinely operated at the Slow Flow Rate setting (14 ul sample/minute), and data acquisition for a single sample typically took 5 min.
Apoptosis assay
Annexin V–fluorescein isothiocyanate (FITC) and propidium iodide (PI) were used to identify and quantify early and late apoptotic cells (Annexin V-FITC Apoptosis Detection Kit, Abcam). Briefly, cells were seeded in a 6-well plate. The adherent and floating cells were combined and washed twice with cold phosphate-buffered saline, resuspended in binding buffer and stained using Annexin V-FITC/PI, according to the manufacturer's instruction. Samples were immediately analyzed by FACS (Accuri C6 Flow Cytometer, Accuri) to differentiate apoptotic cells (Annexin-V positive and PI-negative) from necrotic cells (Annexin-V and PI-positive). Statistical analysis was performed using Accuri C6 flow cytometer software.
Statistical Analysis
Statistical analysis was performed with GraphPad software, and p values were obtained using one-way analysis of variance (ANOVA) followed by post hoc Dunnet. Data are presented as mean ± SEM with the number of independent experiments (run in triplicate). A p-value of p ≤ 0.05 was considered significant.
Acknowledgments
This work was supported by the Pennsylvania Department of Health for Sbarro Health Research Organization (SHRO) and The Ken and Ann Douglas, Charitable Foundation Trust.
References
- Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427-37. doi:10.1038/nrn2151. PMID:17514196
- Jori FP, Napolitano MA, Melone MA, Cipollaro M, Cascino A, Altucci L, Peluso G, Giordano A, Galderisi U. Molecular pathways involved in neural in vitro differentiation of marrow stromal stem cells. J Cell Biochem. 2005;94:645-55. doi:10.1002/jcb.20315. PMID:15547939
- Galderisi U, Jori FP, Giordano A. Cell cycle regulation and neural differentiation. Oncogene. 2003;22:5208-19. doi:10.1038/sj.onc.1206558. PMID:12910258
- Becker EBE, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 2004;72:1-25. doi:10.1016/j.pneurobio.2003.12.005. PMID:15019174
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-93. doi:10.1242/dev.091744. PMID:23861057
- Wang W, Bu B, Xie M, Zhang M, Yu Z, Tao D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009;89:1-17. doi:10.1016/j.pneurobio.2009.01.007. PMID:19619927
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci. 2005;102:8333-8. doi:10.1073/pnas.0500989102. PMID:15923260
- Frade JM, Ovejero-Benito MC. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle. 2015;14:712-20. doi:10.1080/15384101.2015.1004937
- Skovira JW, Wu J, Matyas JJ, Kumar A, Hanscom M, Kabadi SV, Fang R, Faden AI. Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J Neuroinflammation. 2016;13:299.
- Lénárt N, Brough D, Dénes Á. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J Cereb Blood Flow Metab. 2016; Oct 36:1668-85. doi:10.1177/0271678X16662043. PMID:27486046
- Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P. The role of inflammation in perinatal brain injury. Nat Rev Neurol. 2015;11:192-208. doi:10.1038/nrneurol.2015.13. PMID:25686754
- Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. Eur J Neurosci. 2001;13:1101-6. doi:10.1046/j.0953-816x.2001.01474.x. PMID:11285007
- Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide Sensitizes Neonatal Hypoxic-Ischemic Brain Injury in a MyD88-Dependent Manner. J Immunol. 2009;183:7471-7. doi:10.4049/jimmunol.0900762. PMID:19917690
- Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP. Lipopolysaccharide-induced interleukin (IL)-4 receptor-α expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav Immun. 2012;26:766-77. doi:10.1016/j.bbi.2011.10.003. PMID:22024136
- Smith PLP, Hagberg H, Naylor AS, Mallard C. Neonatal peripheral immune challenge activates microglia and inhibits neurogenesis in the developing murine hippocampus. Dev Neurosci. 2014;36:119-31. doi:10.1159/000359950. PMID:24642725
- Bonfill-Teixidor E, Otxoa-de-Amezaga A, Font-Nieves M, Sans-Fons MG, Planas AM. Differential expression of E-type prostanoid receptors 2 and 4 in microglia stimulated with lipopolysaccharide. J Neuroinflammation. 2017;14:3.doi:10.1186/s12974-016-0780-7. PMID:28086956
- Sun A, Bagella L, Tutton S, Romano G, Giordano A. From G0 to S phase: a view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J Cell Biochem. 2007;102:1400-4. doi:10.1002/jcb.21609. PMID:17979151
- Clark AR., Maandag ER, Marian VR, Van Der Lugt NMT, Van Der Valk M, Hooper ML, Berns A, Rielef H Te. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328-30. doi:10.1038/359328a0. PMID:1406937
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295-300. doi:10.1038/359295a0. PMID:1406933
- Lee E-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288-94. doi:10.1038/359288a0. PMID:1406932
- Fong BC, Slack RS. RB: An essential player in adult neurogenesis. Neurogenesis. 2017;4:e1270382.doi:10.1080/23262133.2016.1270382. PMID:28229086
- Lui DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005;19:719-32. doi:10.1101/gad.1296405. PMID:15769944
- Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, McNamara SM, Park DS, Rudnicki M, Slack RS. P107 Regulates Neural Precursor Cells in the Mammalian Brain. J Cell Biol. 2004;166:853-63. doi:10.1083/jcb.200403156. PMID:15353549
- Vanderluit JL, Wylie CA, McClellan KA, Ghanem N, Fortin A, Callaghan S, MacLaurin JG, Park DS, Slack RS. The Retinoblastoma family member p107 regulates the rate of progenitor commitment to a neuronal fate. J Cell Biol. 2007;178:129-39. doi:10.1083/jcb.200703176. PMID:17591923
- Claudio PP, Tonini T, Giordano A. The retinoblastoma family: twins or distant cousins? Genome Biol. 2002;3: reviews3012.doi:10.1186/gb-2002-3-9-reviews3012. PMID:12225593
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220-7. doi:10.1038/sj.onc.1209615. PMID:16936740
- Dyson NJ. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492-502. doi:10.1101/gad.282145.116. PMID:27401552
- Frade JM. Unscheduled re-entry into the cell cycle induced by NGF precedes cell death in nascent retinal neurones. J Cell Sci. 2000;113(Pt 7):1139-48.
- Park KHJ, Hallows JL, Chakrabarty P, Davies P, Vincent I. Conditional Neuronal Simian Virus 40 T Antigen Expression Induces Alzheimer-Like Tau and Amyloid Pathology in Mice. J Neurosci. 2007; 27:2969-78. doi:10.1523/JNEUROSCI.0186-07.2007. PMID:17360920
- Byrnes KR, Faden AI. Role of cell cycle proteins in CNS injury. Neurochem Res. 2007;32:1799-807. doi:10.1007/s11064-007-9312-2. PMID:17404835
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995; 81:323-30. doi:10.1016/0092-8674(95)90385-2. PMID:7736585
- Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10.doi:10.1186/1747-1028-7-10. PMID:22417103
- Fiorentino FP, Symonds CE, Macaluso M, Giordano A. Senescence and p130/Rbl2: a new beginning to the end. Cell Res. 2009;19:1044-51. doi:10.1038/cr.2009.96. PMID:19668264
- Genovese C, Trani D, Caputi M, Claudio PP. Cell cycle control and beyond: emerging roles for the retinoblastoma gene family. Oncogene. 2006;25:5201-9. doi:10.1038/sj.onc.1209652. PMID:16936738
- Indovina P, Marcelli E, Casini N, Rizzo V, Giordano A. Emerging roles of RB family: new defense mechanisms against tumor progression. J Cell Physiol. 2013;228:525-35. doi:10.1002/jcp.24170. PMID:22886479
- Kruman II. Why do neurons enter the cell cycle? Cell Cycle. 2004;3:769-73.
- Ekdahl CT, Claasen J-H, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci. 2003;100:13632-7. doi:10.1073/pnas.2234031100. PMID:14581618
- Monje ML. Inflammatory blockade restores adult hippocampal neurogenesis. Science (80- ). 2003;302:1760-5.
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081-8. doi:10.1038/ncb1629. PMID:17704767
- Das ND, Choi MR, Jung KH, Park JH, Lee HT, Das A, Kim SH, Chai YG. Functional Analysis of Histone Demethylase Jmjd2b on Lipopolysaccharide-Treated Murine Neural Stem Cells (NSCs). Neurotox Res. 2013 Feb;23(2):154-65. doi:10.1007/s12640-012-9346-3. Epub 2012 Aug 14.
- Cacci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L. In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia. 2008;56:412-25. doi:10.1002/glia.20616. PMID:18186084
- Macaluso M, Caracciolo V, Rizzo V, Sun A, Montanari M, Russo G, Bellipanni G, Khalili K, Giordano A. Integrating role of T antigen, Rb2/p130, CTCF and BORIS in mediating non-canonical endoplasmic reticulum-dependent death pathways triggered by chronic ER stress in mouse medulloblastoma. Cell Cycle. 2012;11:1841-50. doi:10.4161/cc.20242. PMID:22544282