
ABSTRACT
Senescence contributes to the local and systemic aging of tissues and has been associated with age-related diseases. Recently, roles for this process during pregnancy have come to light, the dysregulation of which has been associated with adverse pregnancy outcomes such as preterm birth. Here, we summarize recent advances that support a role for senescence in birth timing and propose new aspects of study in this emerging field.
The problem of preterm birth
Preterm birth, defined as delivery before 37 completed weeks of gestation, has been a global health concern for far too long. In the United States, nearly 12% of deliveries are born preterm every year.Citation1 Premature delivery accounts for nearly 75% of early neonatal morbidity and mortality, with 15 million premature births and greater than 3 million stillbirths worldwide.Citation1-5
The coordinated events leading to normal, term parturition has been studied in depth. These include (1) transformation of the myometrium to highly contractile cells, (2) cervical ripening, and (3) weakening and rupture of fetal membranes in response to inflammation of the adjacent decidua,Citation5,Citation6 triggering coordinated myometrial contractility resulting in delivery of the fetus. These processes are associated with decidual and membrane activation, which comprise the anatomical and biochemical events towards the withdrawal of decidual support for pregnancy, separation of the chorioamniotic membranes from the decidua and eventually, membrane rupture.Citation7,Citation8 The transition from quiescence to activation is characterized by a functional progesterone withdrawal in the setting of unchanged serum progesterone levels, resulting from epigenetic control of progesterone signaling and reduced responsiveness, increased estrogen effects, and increased contraction-associated protein (CAP) expression in the myometrium, including oxytocin and gap junction protein connexin 43, resulting in a proinflammatory milieu towards coordinated uterine contractions.Citation5,Citation7,Citation9,Citation10
Mechanisms underlying preterm birth remain unclear. Risk factors towards preterm delivery are wide-spanning and involve gene-gene and gene-environment interactions. It is unknown whether preterm birth harnesses the same pathways as normal delivery. Variables that contribute to this multifactorial disorder include genetic predisposition (family history), infection/inflammation, environmental inducers of oxidative stress (such as cigarette smoking), short cervix, progesterone resistance, extremes in maternal age (young and old), assisted reproductive technology, stretch signaling originating from multiple pregnancy, short inter-pregnancy interval, socioeconomic factors resulting in late to no prenatal care, and prior preterm birth.Citation1,Citation5,Citation7,Citation11
In spite of our best efforts, current strategies to prevent preterm birth have been limited. Despite years of investigation to identify strategies against preterm birth, the most common interventions implemented for the prevention or treatment preterm labor include bed rest, tocolytics, antibiotic treatment, and cervical cerclage. These methods have proved to be of little or no benefit in women with multiple risk factors.Citation12 Progesterone supplementation for women with particular risk factors such as previous preterm delivery or a short cervix has shown promise but has not shown improvement in women with other or additional risk factors.Citation13,Citation14
Further studies are required to study risk factors, singularly or in combination, to determine whether signaling pathways respond to risk factors differentially. This will help to differentiate preterm birth into subtypes based upon etiology and molecular signature.Citation5,Citation7 Our limited understanding of the mechanism(s) underlying preterm birth and parturition has resulted in failure to effectively detect and apply successful treatments or interventions.Citation5,Citation12
These signaling pathways are thought to ultimately converge upon the activation of fetal and maternal tissues to stimulate myometrial contractions and/or rupture of the fetal membranes. In recent years, cellular senescence of maternal-fetal tissues has been proposed as a mechanism and common pathway integrating multiple risk factors for birth timing. This article will review our current understanding of this process in pregnancy events.
Basic concepts of senescence
Senescence is a process by which cells enter a state of durable growth arrest and irreversibly cease proliferation without undergoing cell death (apoptosis). This process was first described by Hayflick and Moorhead in cell culture who identified intrinsic factors that could limit cellular proliferation in normal cells.Citation15,Citation16 They speculated that a molecular “stopwatch” limiting proliferation and growth could regulate organismal aging.
Later studies showed that senescence occurs in response to a range of physiological stresses and molecular damage when a cell is under duress.Citation17 These stress signals include genotoxic stress in the forms of unrepaired DNA damage or telomere loss, oxidative stress, and uncontrolled oncogenic signaling. Senescence has since been identified in multiple metabolically active tissues to halt cell proliferation and elicit local inflammatory responses to stimulate macrophage recruitment for cell clearance and tissue remodeling.Citation18,Citation19 Its involvement has been reported extensively as the body's protective response against malignant growth but also in normal physiological processes such as megakaryocyte maturation,Citation20 hepatic growth,Citation21 cellular aging,Citation22 and recently, in embryonic development and patterning.Citation23,Citation24 Whole body, organismal aging has also been shown to be associated with increased numbers in DNA mutationsCitation25,Citation26 and accumulation of senescent cells in aged tissues, diminishing the regenerative properties of progenitor cells.Citation19,Citation27,Citation28
As opposed to apoptosis, which is a controlled, programmed cell death necessary for the homeostasis of multicellular organisms, senescence, a form of biological aging, is a terminal differentiation with reduced functional capacity of the cell. This is characterized by changes in the cell's gene and protein expression to influence the metabolic capacity of cells around it, thus changing the extracellular environment to limit a tissue's potential for renewal.
All senescent cells appear to exhibit a flat, vacuolated morphology, with increased senescent-associated beta-galactosidase (SA-β-gal) staining associated with the accumulation of lysosomes.Citation29 However, its molecular signature varies with the specific trigger for senescence. Molecular marks used to describe senescent cells include telomere shortening;Citation30 accumulation of DNA damage and derepression of the INK4a/ARF locus;Citation31 dysregulation by tumor suppressors p53 and retinoblastoma protein (Rb);Citation18 markers of heterochromatin such as SAHF at H3K9 methylation;Citation32 altered levels of cell cycle regulators p21, p16, p19, and HP1γ;18 decreased expression of nuclear lamin B1,33 loss of nuclear HMGB1 and its secretion into the extracellular milieu,Citation34 and enhanced unfolded protein response aka proteostasis,Citation35 among others.
Telomeres are short, tandemly repeated DNA elements at the ends of linear chromosomes that protect the DNA ends from degradation and/or recombination.Citation30,Citation36,Citation37 DNA replication machinery is unable to copy the ends of linear molecules, so telomeres become progressively shorter (telomere attrition) with every round of cell division.Citation38 Telomere attrition has been implicated in cellular aging and aging-related diseases, and factors that may decrease longevity, including stress or obesity, have been reported to decrease telomere length.Citation39,Citation40 Critically-short or dysfunctional telomeres can elicit a DNA damage response to initiate senescence or apoptosis and are marked with phosphorylated histone H2AX, or γH2AX.Citation41 Proteins that comprise the shelterin complex normally protect the shortened ends from p53-mediated DNA repair.Citation42
The INK4a/ARF locus encodes two tumor suppressors, p16INK4a and ARF, via different reading frames, both of which are important inducers of cellular senescence.Citation43 While p16INK4a inhibits the activity of cyclin-dependent kinases CDK4 and CDK6 by imposing a G1 cell cycle arrest, ARF enhances p53 activity and stability through inactivation of the p53-degrading ubiquitin ligase MDM2.31,44 Expression of the INK4a/ARF locus is normally very low to undetectable in most tissues in young organisms but increases with chronological aging:45 the expression of p16INK4a and ARF markedly increase with aging in rodent tissues and have been shown to increase with aging in human kidney and skin cells.Citation45-48 Mice deficient in p16INK4a have increased regeneration potential of stem cells,Citation45,Citation49,Citation50 suggesting a contribution of p16INK4a to the age-associated decline in tissue regeneration potential of mammalian aging. While the mechanisms responsible for the increased expression of p16INK4a in aging tissues are not completely understood, an association with Polycomb and homeobox gene activity have also been proposed.Citation51 The expression of the INK4a/ARF locus can also be accelerated by high oncogenic activity, a phenomenon that has been called oncogene-induced senescence, or OIS.Citation52,Citation53 Protection from carcinogenesis by activating the INK4a/ARF locus has been extensively described elsewhere.Citation31,Citation44
Senescence associated secretory phenotype
Two hallmarks of cellular senescence are an irreversible arrest of cell proliferation and the development of a pro-inflammatory and pro-tumorigenic local environment, which is now referred to as senescence-associated secretory phenotype (SASP) aka immuno-senescence.Citation17,Citation54 Although senescent cells are no longer mitotic, they remain metabolically active and capable of shaping the secretory cytokine milieu to influence the local microenvironment. This explains how a small number of senescent cells can promote aging phenotypes and pathologies via local and systemic effects. SASP was first described as an entity in 2001 when it was discovered that senescent human fibroblasts could stimulate neighboring premalignant and malignant cells to proliferate in culture and form tumors in mice.Citation55,Citation56 However, multiple prior works had already identified the synergistic effect of the altered microenvironment to enable premalignant cells to become fully malignant.Citation57-59
SASP entails the secretion of a collection of potent biological factors that promote inflammation, invasion, angiogenesis, and ironically, cell proliferation on neighboring cells and the surrounding tissue.Citation17 Several constituents comprising SASP have been identified to date: cytokines, chemokines, MMP's, and constitutents of the TGFβ, IGF, and VEGF signaling pathways.Citation18,Citation60 Recently, attention has also been focused on extracellular vesicles as conveyors of senescence signals outside the cell.Citation61 These composite mediators create a microenvironment that facilitates the progression of senescent cells, induces proliferation or epithelial-to-mesenchymal transformation and invasion in neighboring cells, and reinforces the senescence growth arrest through autocrine and paracrine mechanisms.Citation24,Citation54,Citation62,Citation63
SASP involvement in parturition and preterm delivery has been proposed since there is overlap in the SASP signature with factors known to stimulate coordinated myometrial contractility, particularly pro-inflammatory cytokines and prostaglandins. In addition to local paracrine effects, senescence of maternal-fetal tissues appear to have a mechanical contributory role towards delivery: senescence of maternal tissues weaken the decidual anchor of the placenta to the mother while senescence in the fetal membranes weaken this barrier in preparation for rupture. Furthermore, triggers for cellular senescence such as DNA damage and oxidative stress are also known to be risk factors for preterm birth, making this an alluring hypothesis.
Senescence in reproductive tissues
Placental senescence
The placenta is an embryonically-derived, discoid organ that serves as the interface between the fetus and the mother, providing functions necessary for fetal survival, growth, and developmentCitation64. Grossly, it is comprised of the fetal placenta which is genetically identical to the fetus, and the maternal decidual basalis, which developed from maternal stromal tissue (see below: “Decidual senescence”). Its functions are numerous, including nutrient uptake, waste elimination, gas exchange, thermo-regulation of the fetus, barrier against infection, hormonal production, among others.Citation64
Placental “aging” has been implicated in functional insufficiency to meet the needs of the fetus and thus compromise fetal viability,Citation65 resulting in abnormal pregnancy outcome. However, this concept has not been mechanistically investigated. Placental aging has been correlated with women with advanced maternal age in the form of increased gross and histologic calcificationCitation66 and accumulation of lipofuscin pigmentsCitation67 and fibrinoid material comparable to amyloid.Citation68 Sonographic changes during late gestation were suspected to reflect decline in placental function secondary to physiological stress rather than normal maturational development.Citation69 Indeed, pregnancy itself can impart stress (oxidative, hypoxic and nitrative stress) to alter placental development. These stressors can be further exacerbated by smoking, intrauterine growth restriction (IUGR), preeclampsia or in miscarriage.Citation65,Citation70
While previous studies have denoted physiological “senescence” to be synonymous with aging and functional decline, placental senescence in the molecular sense has only recently been investigated. Several older studies denote senescence/aging to encompass placental apoptosis which increases with pregnancy progression.Citation71 Molecular studies have identified placental apoptosis in the setting of either reduced or unchanged telomerase activity.Citation72-74 While placental apoptosis has been deemed normal for the formation of villous trophoblast bilayer and syncytiotrophoblast formation, it is yet unclear whether cellular senescence in the placenta itself is implicated in pathologic pregnancies such as preeclampsia, preterm birth or IUGR. Recently, studies have shown that fetal growth restriction is associated with telomere shortening and increased expression in cell senescence markers such as p21, p16 and elongation factor 1α in decidual-placental samples taken near the umbilical cord.Citation75 Further studies are warranted to investigate whether (1) the positive staining was in the decidua vs. placenta or both; (2) placental senescence itself exists independently of placental maturation, and if so, (3) whether senescence can induce pathological pregnancy states.
The concept of placental aging has been controversial. Indeed, when the concept of placental aging is tested against the definition of true aging (progressive, irreversible loss of functional capability) versus maturational, time-related changes (with increased functional capacity), it elicits more questions than answers.Citation71 Placental senescence in its truest sense would be intriguing, since the embryo and its placenta represent genetically identical tissues which age at different rates.Citation76 The focal thinning of the villous syncytiotrophoblast found in terminal villi has often been cited as evidence of syncytial senescence however these thinned areas are optimally adapted to increase trophoblastic surface area and facilitate gas transfer.Citation77 Placentae from women with severe pre-eclampsia appear more mature for the length of gestation and has been classed as “premature aging” but pathologically would be more accurately regarded as accelerated maturation to increase the diffusion capacity of the placenta in the setting of an adverse maternal environment.Citation71 Although placental growth does slow in the last few weeks of gestation, its growth does not cease and trophoblast cells can proliferate, repair and replace cells in unfavorable maternal milieu or ischemic damage.Citation78,Citation79
Decidual senescence
The maternal decidua is a transiently-lived, terminally-differentiated organ that begins to form immediately after embryo implantation. During the attachment phase of implantation in rodents, the physical and molecular interaction of the embryonic trophectoderm with the uterine epithelium initiates differentiation of the underlying stromal cells (decidualization). The role of decidual stromal cells has been posited to direct placental development and nourish the developing embryo as a readily-available glycogen source released upon phagocytosis by the trophoblast as the nascent placenta develops.Citation80-82 The decidua acquires an epithelioid phenotype with upregulation of epithelial markers such as ZO1 and cadherins; thus, it is also presumed to slow the progression of trophoblast invasion, as unopposed invasion would be detrimental to maternal health.Citation80,Citation81,Citation83,Citation84 The decidua remains in close proximity to the chorion/amnion and is the maternal component of the fetal-maternal interface for the remainder of pregnancy. In rodents, decidualization is maximal on day 8, then ceases to proliferate and thins to accommodate the growing fetus for the remaining duration of pregnancy.
In line with its supportive role, the terminal differentiation of both decidual stromal cells and interspersed trophoblast giant cells results in marked cellular polyploidy, which is a widespread physiological phenomenon and is part of the normal developmental program for the formation of highly differentiated cells, including megakaryocytes, cardiomyocytes, Purkinje fibers, retinal ganglion cells, and hepatocytes.Citation20 Polyploid cells in each tissue type can form in response to stress or injury, or if unscheduled, can trigger carcinogenesis by inducing chromosomal instability. Polyploid cells can form by various mechanisms, including cell fusion (osteoclasts, skeletal muscles), endomitosis (megakaryocytes) and endoreduplication (proposed for decidual cells and trophoblast giant cells, in which cells alternate S and G phases without undergoing mitosis). Whatever the mechanism driving polyploidy, the increase in cellular DNA content leads to increased synthetic capacity. Notably, polyploidy has also been associated with senescence and SASP.Citation85
The contribution of decidual aging in birth timing has been recently reported. A genetic mouse model of spontaneous preterm birth was developed harboring a conditional deletion of tumor suppressor p53 in uterine tissues (uterine Trp53 deletion = p53d/d dams).Citation8,Citation86-89 Notably, p53 activity diminishes in mice as they age,Citation90 and decreased Trp53 expression with terminal differentiation is observed in developing organs in mouse embryos.Citation91
p53d/d dams have normal ovulation, fertilization, and implantation but found to have decidual growth restriction and increased decidual polyploidy compared to control littermatesCitation8. Intriguingly, p53d/d dams had a strong predilection towards preterm delivery, with 50% of females exhibiting spontaneous preterm delivery with 100% stillbirth, compared to none in control littermates.Citation8 Post-implantation decidual cells in p53d/d dams showed premature terminal differentiation and senescence-associated growth restriction with increased levels in p21, pAkt, cyclooxygenase (COX)-2, and prostaglandin F synthase (PGFS)/prostaglandin (PG)F2a. This group showed that premature decidual senescence in p53d/d dams was secondary to heightened mammalian target of rapamycin (mTORC1) signaling, as determined by increased levels of phospho-S6. When pregnant females were fed intermittent doses of rapamycin (a selective mTORC1 inhibitor), they demonstrated reduced decidual mTORC1 activity and decidual growth restriction, normalized progression of decidual senescence, and the prevention of preterm birth in this susceptible model.Citation86
Due to the strong association of infection/inflammation with preterm birth, p53d/d dams were exposed to LPS to evaluate their susceptibility to inflammatory insults. Indeed, even a low dose of ultrapure lipopolysaccharide (LPS; 10μg), which had no effect on wild type dams, resulted in preterm birth in 100% of p53d/d females assessed. Pretreatment with mTORC1 inhibitor with progesterone supplementation prevented preterm birth in p53d/d females without notable adverse effects to either mother or pups in treated control or p53d/d females.Citation87 Later studies showed that pretreatment with metformin, an antidiabetic agent with mTORC1 inhibitory and AMPK activating properties, also showed protection against preterm birth in combination with progesterone.Citation88 These results show that targeting gene-environment interactions by combination therapy with mTORC1 inhibition with progesterone supplementation could prevent preterm birth.
Of note, historical studies have shown that decidualization is less robust in aged mice and associated with poor pregnancy outcome.Citation92,Citation93 Intriguingly, the limited decidual cell response in aged mice has been reported to be partially overcome by ovariectomy or caloric retriction, implicating mTOR-mediated activity prior to molecular evidence.Citation94
The accumulation of senescent decidual cells results in enhanced local SASP, which can stimulate myometrial contractility, fetal membrane activation and weakening in the presence of proinflammatory cytokines and mechanical weakening with enhanced of myometrial contractility. Indeed the progression of decidual senescence was noted in WT dams in term pregnancy, but at an accelerated rate in p53d/d females.Citation86 What is more, treatment of primary decidual cells in culture with an mTORC1 inhibitor plus progesterone limited the activity of COX-2 and reduced SASP-associated and labor-promoting cytokines IL-6 and IL-8.87 Proteomics analysis in p53d/d deciduae showed downregulation of a cluster of antioxidant enzymes, suggesting increased oxidative stress in these mice as a cause of preterm birth.Citation89 Finally, increased decidual senescence was shown to be present in preterm laboring women in a Japanese cohort compared to term women.Citation87 Collectively, these data provide support for a role of decidual senescence in driving preterm labor.
It is intriguing that, in contrast to its role in tumorigenesis, the absence of uterine p53 results in heightened senescence mediated by mTOR signaling.Citation60 Previous reports have shown that p53 inhibits mTOR signalingCitation95,Citation96 and, in recent reports, Trp53 deletion appears to paradoxically drive senescensce via increased mTORC1 signaling in a human fibrosarcoma cell line.Citation97 Roles for p53 in development such as during embryogenesis or in reproduction, which can reactivate developmental pathways in a controlled fashion, may differ from its well-described roles in cancer with uncontrolled proliferation. These data, along with our present observations, suggest differential roles and regulation of p53 between physiologic (adult and embryonic) and pathophysiologic (malignant) systems.
Senescence in fetal membranes
Juxtaposed to an outer layer of maternally-derived decidual stromal cells, the fetal membranes are comprised principally by the embryonically-derived inner amnion and outer chorion.Citation98 The membranes are not vascularized, thus limiting the ability to transfer nutrients between mother and fetus.Citation6 While both the amnion and chorion can synthesize prostaglandins (PG's), the chorion expresses 15-hydroxyprostaglandin dehydrogenase (PGDH), which inactivates bioactive PGs and blocks amniotic PGs from accessing the myometrium.Citation6 The initiation of parturition is associated with decreased PGDH activity in the chorion to allow PG's to reach the myometrium. Fetal glucocorticoid action can stimulate COX-2-mediated PG production while cortisol can inhibit PGDH expression/activity to coordinate maternal-fetal interactions to initiate parturition.Citation6,Citation99 Recent work in mice has also illuminated the contribution of embryo-derived surfactant to signal birth timing, suggesting that embryonic signals help coordinate birth timing.Citation99
In most term pregnancies in women, uterine contractions precede fetal membrane rupture. It is thought that uterine contractions mechanically weaken the fetal membranes in preparation for delivery.Citation6 This process involves matrix remodeling via MMP's and fetal membrane apoptosis. Typically, the choriodecidua ruptures first, followed by the amnion.Citation6 However, this sequence is not stereotypical, with nearly 10% of women having fetal membrane rupture prior to contractions (e.g., PPROM). Fetal membrane apoptosis has been associated with infection/inflammation, cigarette smoking, uterine overdistension, decidual hemorrhage, and genetic predisposition.Citation100
In addition to progressive apoptosis, senescence has also been identified in the fetal membranes prior to delivery. To evaluate fetal membrane senescence in vivo, term pregnant WT mice were evaluated for cellular senescence by SA-β-gal staining and for p53 and MAPK/p38 activity.Citation101 Fetal membranes were shown to senesce from day 12 of pregnancy until day 18. Decidual senescence was also evaluated in these mice and was present from day 10 until day 15–18. These studies showed active p53 on day 18 on fetal membranes, while no change was observed in phosphorylated p53 from placental and decidual/uterine samples. It would be interesting to evaluate for apoptosis as well, since p53 is known to induce apoptosis in the setting of oxidative or genotoxic stress.
Senescence of primary amniotic cells in vitro was evaluated after exposure to oxidative stress with cigarette smoke extract, which showed activation of ASK1, P-p38 MAPK and p19(Arf) which correlated with percentage of SA-β-gal-positive cells.Citation102 Telomere length in fetal leukocytes and placental membrane cells were also shown to be shortened in tissues from preterm babies compared to those from term babies, implicating telomere shortening as a surrogate for oxidative stress and senescence.Citation103 Thus, further study is warranted to evaluate the contribution of senescence in fetal membranes in preterm birth and across different etiologies of preterm delivery.
It has been proposed that labor signaling originates from the fetal membranes via increased fetal membrane senescence.Citation104 This process is thought to activate the decidua to secrete proinflammatory cytokines and PGs, triggering a pathway towards myometrial contractility for labor. The initiating factor to cause fetal membrane senescence remains to be determined; proposed initiators include cell free DNA, telomere shortening, soluble HMGB1.Citation105,Citation106 Notably, patients with high levels of cell free DNA in the mid-trimester are at increased risk for spontaneous preterm delivery.Citation107
So far, clinical markers for placental/fetal membranes aging as a risk factor of preterm birth or PPROM have not been identified. Interestingly, in p53d/d females presented above, senescence in fetal membranes was identified at similar degrees in both control and p53d/d mice, suggesting that its contribution did not significantly influence preterm birth phenotype in this model. Notably, placental senescence was not identified in p53d/d females, suggesting that placental aging was not influenced by decidual senescence. Nonetheless, the contribution of such membrane aging to parturition timing and interplay with decidual senescence is an exciting field and warrants further study.
Future perspectives
In this article, we summarize recent reports highlighting a potential role for cellular senescence in birth timing. Recent findings show that (1) cellular senescence is present in both fetal and maternal tissues and accumulates as pregnancy approaches delivery, and (2) that the premature progression of senescence and SASP may contribute to the myometrial contractility and fetal membranes-decidual activation seen in preterm birth in mice and women.Citation8,Citation82,Citation86,Citation106,Citation108,Citation109
The concept of molecular senescence contributing to the timing of labor and delivery was identified in murine models as early as 20108. In mice, the authors identified that the decidua undergoes progressive cellular senescence with SA-β-gal staining and noted that p53d/d dams showed accelerated senescence; no difference in senescence was noted in the fetal membranes in this model and SA-β-gal staining was not evident in placental tissues, suggesting that decidual senescence was the primary driver of preterm birth in this model. These studies also suggest that environmental factors that accelerates the rate of senescence (e.g. infection, inflammation, environmental insults) to limit decidual growth and lifespan can result in preterm delivery, thus showing that gene-environment interactions underlie preterm delivery. Proof of principle studies using this animal model exhibiting accelerated decidual senescence, spontaneous preterm birth and heightened sensitivity to LPS-triggered preterm birth showed that premature senescence could be targeted (both by pharmacologic and genetic means) to prevent preterm birth.Citation87,Citation88
Additional studies have identified fetal membrane senescence in in vitro studies and in normal rodent parturition.Citation103 As the decidua and fetal membranes age and undergo cellular senescence, local SASP is enhanced with secretion of proinflammatory cytokines, soluble factors, and prostaglandins to achieve a senescence threshold which can trigger a common pathway of fetal membranes activation and myometrial contractility ( and ). This observation would be a paradigm shift in our understanding of preterm birth.Citation110
Figure 1. A scheme depicting potential contribution of decidual mTORC1 and senescence in setting the parturition clock. Term pregnancy encompasses progressive decidual senescence over the course of pregnancy (pink arrow) until a threshold is encountered, triggering PG synthesis, myometrial activation and contractility, culminating in birth. In the mouse model of preterm labor in which Trp53 is conditionally deleted in the uterus, decidual senescence begins prematurely due to activation of an pAkt-mTORC1-p21-Cox2 signaling axis and reaches the senescence threshold in a shorter gestational time frame, leading to preterm delivery. Several risk factors of preterm birth, including genetics, stress and inflammation/infection have been shown to contribute to the senescence process in other systems, and we speculate that these factors pathologically push decidual senescence toward the threshold. Furthermore, normal parturition involves cervical ripening along with myometrial activation in preparation for parturition, which does not occur in the p53d/d females: these females exhibit dystocia and stillbirth. Rapamycin, an inhibitor of mTORC1 signaling, can attenuate premature decidual senescence seen in p53d/d females and rescue preterm labor (Adapted from Citation110).
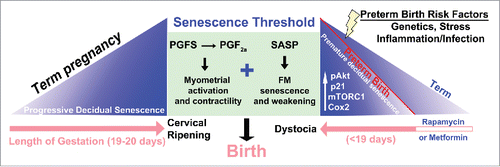
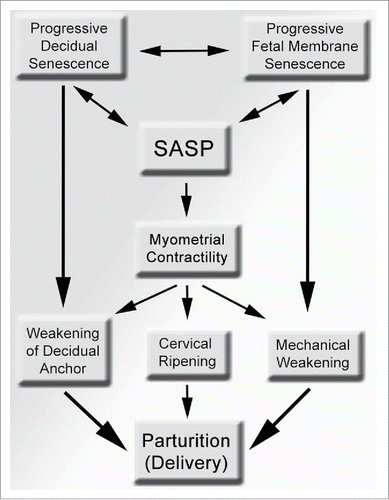
Figure 2. A scheme showing the potential contributions of senescence in various intrauterine tissue compartment to the process of parturition and preterm birth. Progressive senescence in the maternal decidua can structurally weakens its anchoring role to the mother, while senescence in the fetal membranes results in mechanical weakening and can lead to rupture of membranes. Myometrial contractility can also promote decidual and fetal membrane weakening via mechanical shear stress, which can be heighted by local, intrauterine SASP, resulting in parturition. SASP, senescence-associated secretory phenotype.
While the implications of these preliminary studies are alluring, further investigation to their relevance across human populations is required. We have previously shown that in mice, normal parturition is correlative with a progressive accumulation of decidual senescence,Citation86 but this phenomenon has not yet been investigated in depth in women. Increased decidual senescence was noted in preterm laboring women in a Japanese cohort compared to term women.Citation87 However, do these findings extend into other populations with heterogenous backgrounds and different exposures? Is it truly a driver of preterm delivery or merely a consequence after labor signaling has begun? Is it associated with a progression of a normal physiological process in women, or is its presence pathological? Do several processes converge on senescence to attain a threshold of senescence or is labor the additive result of parallel pathways? If it is the result of converging processes, can we target senescence as a preventative measure early in pregnancy in high risk women or even in women with no risk factors? Establishing placental biobanks to study the degree of senescence by molecular markers in laboring versus medically indicated, Caesarian section-derived preterm tissues will help elucidate the incidence and contribution of this process to labor across populations.
These murine studies also raise the question for the role of p53 itself in preterm birth. Do women with higher risk of preterm birth have genetic mutations or SNPs in p53 or the mTOR pathway? Increased gH2AX and polyploidy seen in p53d/d decidua suggest heightened genotoxic stress and increased oxidative stress as profiled by proteomicsCitation89. These suggest a central feature of genotoxic and oxidative stress-induced senescence in the decidua underlying preterm birth phenotype. Genetic and molecular screens in women with preterm birth to evaluate for the presence of mutations or oxidative stress will require further investigation and benefit from the establishment of placental biobanking.
Proof-of-principle studies in rodents show that the inhibition of mTORC1 signaling with rapamycin or metformin has slowed the progression of senescensce, improved decidual health and allowed pregnancy to complete to term without negative effects on neonatal pups. Metformin has already been deemed safe for clinical use during human pregnancy, and investigation towards repurposing this relatively inexpensive drug in pregnancy is feasible. It remains to be seen whether these compounds can be safe for use in pregnancy and can inhibit decidual and fetal membrane aging for prematurity prevention. Once molecular characterization of this cellular process in pregnancy is established, it will become feasible to envisage preterm birth as a collection of disorders and stratify cases by molecular signature to target the cellular processes that ultimately trigger preterm birth rather than attempting to stop labor once it has begun. By doing so, we may potentially identify biomarkers to develop novel tests or employ predictive calculators to help stratify women at risk of delivering preterm with known risk factors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Contributions
J.M.C. and D.M.A. wrote the manuscript.
Acknowledgments
We regret that space limitations precluded us from citing many relevant references. We would like to thank S.K. Dey for helpful discussions and the Vanderbilt PreCitation3 initiative for their support.
References
- March of Dimes, PMNCH, Save the Children, WHO. Born too soon: The global action report on preterm birth. Eds. Howson CP, Kinney MV, Lawn JE. Geneva, Switzerland: World Health Organization, 2012.
- Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75-84. PMID:18177778
- Lawn JE, Blencowe H, Waiswa P, Amouzou A, Mathers C, Hogan D, Flenady V, Froen JF, Qureshi ZU, Calderwood C, et al. Stillbirths: rates, risk factors, and acceleration towards 2030. Lancet. 2016;387:587-603. PMID:26794078
- Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, Rudan I, Campbell H, Cibulskis R, Li M, et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012;379:2151-61. PMID:22579125
- Rubens C, Sadovsky Y, Muglia L, Gravett M, Lackritz E, Gravett C. Prevention of preterm birth: Harnessing science to address the global epidemic. Science Translational Medicine. 2015;6:262sr5. PMID: 25391484
- Mesiano S, DeFranco E, Muglia LJ. Parturition. In: Tony M. Plant and Anthony J. Zeleznik, editors. Knobil and Neill's physiology of reproduction. Elsevier Science, 2015(2):1875-1925.
- Romero R, Dey SK, Fisher S. Preterm labor: One syndrome, many causes. Science. 2014;345:760-5. PMID:25124429
- Hirota Y, Daikoku T, Tranguch S, Xie H, Bradshaw H, Dey SK. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J Clin Invest. 2010;120:803-15. PMID:20124728
- Renthal NE, Williams KC, Montalbano AP, Chen CC, Gao L, Mendelson CR. Molecular regulation of parturition: a myometrial perspective. Cold Spring Harb Perspect Med. 2015;5: a023069. PMID: 26337112
- Hirota Y, Cha J, Dey SK. Revisiting reproduction: Prematurity and the puzzle of progesterone resistance. Nat Med. 2010;16:529-31. PMID:20448578
- Preterm birth: Causes, conequences, and prevention. In: (US) TNAIoM, editor. Committee on understanding premature birth and assuring healthy outcomes. Washington (DC): National Academies Press, 2007.
- Muglia L, Katz, M. The Enigma of Spontaneous Preterm Birth. N Engl J Med. 2010;362:529-35. PMID:20147718
- Meis PJ, Klebanoff M, Thom E, Dombrowski MP, Sibai B, Moawad AH, Spong CY, Hauth JC, Miodovnik M, Varner MW, et al. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348:2379-85. PMID:12802023
- Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH, Fetal Medicine Foundation Second Trimester Screening G. Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med. 2007;357:462-9. PMID:17671254
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585-621. PMID:13905658
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614-36. PMID:14315085
- Wiley C, Campisi, J. From ancient pathways to aging cells— connecting metabolism and cellular senescence. Cell Metabolism. 2016;23:1013-21. PMID:27304503
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729-40. PMID:17667954
- Serrano M. The inflammTORy powers of senescence. Trends Cell Biol. 2015;25:634-6. PMID:26471225
- Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556-66. PMID:23849927
- Gentric G, Desdouets C, Celton-Morizur S. Hepatocytes polyploidization and cell cycle control in liver physiology. Int J Hepatol. 2012; 2012:282430. PMID:23150829
- Campisi J. Aging and cancer cell biology, 2007. Aging Cell. 2007;6:261-3. PMID:17517036
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez, G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013;55(5):1104-18. PMID: 24238962
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells M, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes W. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119-30. PMID:24238961
- Dolle ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci U S A. 2000;97:8403-8. PMID:10900004
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001; 98:10469-74. PMID:11517304
- Llanos S, Serrano M. Senescence and cancer: In the name of immunosuppression. Cancer Cell. 2016;30:507-8. PMID:27728798
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37-42. PMID:26738589
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363-7. PMID:7568133
- Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193-8. PMID:26785477
- Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667-77. PMID:16921403
- Bao X, Wu H, Zhu X, Guo X, Hutchins AP, Luo Z, Song H, Chen Y, Lai K, Yin M, et al. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res. 2015;25:80-92. PMID:25512341
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066-75. PMID:22496421
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613-29. PMID:23649808
- Martínez G, Duran-Aniotz C, Cabral-Miranda F, Vivar JP, Hetz C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell. 2017;16(4):615-623. PMID: 28436203
- Chan SW, Chang J, Prescott J, Blackburn EH. Altering telomere structure allows telomerase to act in yeast lacking ATM kinases. Curr Biol. 2001;11:1240-50. PMID:11525738
- De Lange T. Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol. 2005;70:197-204. PMID:16869754
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611-22. PMID:16136653
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312-5. PMID:15574496
- Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366:662-4. PMID:16112303
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194-8. PMID:14608368
- Schmutz I, de Lange T. Shelterin. Curr Biol. 2016;26:R397-9. PMID:27218840
- Matheu A, Pantoja C, Efeyan A, Criado LM, Martin-Caballero J, Flores JM, Klatt P, Serrano M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004;18:2736-46. PMID:15520276
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265-75. PMID:17055429
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299-307. PMID:15520862
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649-59. PMID:9393858
- Chkhotua AB, Gabusi E, Altimari A, D'Errico A, Yakubovich M, Vienken J, Stefoni S, Chieco P, Yussim A, Grigioni WF. Increased expression of p16(INK4a) and p27(Kip1) cyclin-dependent kinase inhibitor genes in aging human kidney and chronic allograft nephropathy. Am J Kidney Dis. 2003;41:1303-13. PMID:12776284
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379-89. PMID:16911562
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421-6. PMID:16957735
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448-52. PMID:16957738
- Martin N, Popov N, Aguilo F, O'Loghlen A, Raguz S, Snijders AP, Dharmalingam G, Li S, Thymiakou E, Carroll T, et al. Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK(4)a regulation. EMBO J. 2013;32:982-95. PMID:23455154
- Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472-6. PMID:16723993
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593-602. PMID:9054499
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118. PMID:20078217
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072-7. PMID:11593017
- Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117-26. PMID:17409418
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449-65. PMID:2684393
- Rinehart CA, Torti VR. Aging and cancer: the role of stromal interactions with epithelial cells. Mol Carcinog. 1997;18:187-92. PMID:9142212
- DePinho RA. The age of cancer. Nature. 2000;408:248-54. PMID:11089982
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853-68. PMID:19053174
- Urbanelli L, Buratta S, Sagini K, Tancini B, Emiliani C. Extracellular vesicles as new players in cellular senescence. Int J Mol Sci. 2016;17(9):pii: E1408. PMID:27571072
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973-9. PMID:19597488
- Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485-96. PMID:15657080
- Sadovsky YaJ, T. Placenta and placental transport function. In: Tony M. Plant AJZ, editors. Knobil and Neill's Physiology of Reproduction. London: Academic Press, 2015:741-82.
- Sultana Z, Maiti K, Aitken J, Morris J, Dedman L, Smith R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am J Reprod Immunol. 2017;77:e12653. PMID:28240397
- Fujikura T. Placental calcification and maternal age. Am J Obstet Gynecol. 1963;87:41-5. PMID:14056185
- Parmley TH, Gupta PK, Walker MA. “Aging” pigments in term human placenta. Am J Obstet Gynecol. 1981;139:760-6. PMID:7211984
- Burstein R, Frankel S, Soule SD, Blumenthal HT. Aging of the placenta: autoimmune theory of senescence. Am J Obstet Gynecol. 1973;116:271-6. PMID:4145061
- Fisher CC, Garrett W, Kossoff G. Placental aging monitored by gray scale echography. Am J Obstet Gynecol. 1976;124:483-8. PMID:1258904
- Myatt L. Placental adaptive responses and fetal programming. J Physiol. 2006;572:25-30. PMID:16469781
- Fox H. Aging of the placenta. Arch Dis Child Fetal Neonatal Ed. 1997;77:F171-5. PMID:9462184
- Axt R, Meyberg R, Mink D, Wasemann C, Reitnauer K, Schmidt W. Immunohistochemical detection of apoptosis in the human term and post-term placenta. Clin Exp Obstet Gynecol. 1999;26:56-9. PMID:10459437
- Kudo T, Izutsu T, Sato T. Telomerase activity and apoptosis as indicators of ageing in placenta with and without intrauterine growth retardation. Placenta. 2000;21:493-500. PMID:10940199
- Chen RJ, Chu CT, Huang SC, Chow SN, Hsieh CY. Telomerase activity in gestational trophoblastic disease and placental tissue from early and late human pregnancies. Hum Reprod. 2002;17:463-8. PMID:11821296
- Davy P, Nagata M, Bullard P, Fogelson NS, Allsopp R. Fetal growth restriction is associated with accelerated telomere shortening and increased expression of cell senescence markers in the placenta. Placenta. 2009;30:539-42. PMID:19359039
- Parmley T. Placental senescence. Adv Exp Med Biol. 1984;176:127-32. PMID:6496213
- Mayhew TM, Jackson MR, Boyd PA. Changes in oxygen diffusive conductances of human placentae during gestation (10-41 weeks) are commensurate with the gain in fetal weight. Placenta. 1993;14:51-61. PMID:8456089
- Jackson MR, Mayhew TM, Boyd PA. Quantitative description of the elaboration and maturation of villi from 10 weeks of gestation to term. Placenta. 1992;13:357-70. PMID:1438084
- Sands J, Dobbing J. Continuing growth and development of the third-trimester human placenta. Placenta. 1985;6:13-21. PMID:3991472
- Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet. 2001;2:538-48. PMID:11433360
- Finn C. PD. The Uterus. 1975.
- Cha J, Sun X, Dey SK. Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012;18:1754-67. PMID:23223073
- Cha J, Lim H, Dey SK. Embryo implantation. In: Knobil and Neill's physiology of reproduction. Eds. Tony M. Plant and Anthony J. Zeleznik. Elsevier Science, 2015(2):1697-1739.
- Schleich AB, Frick M, Mayer A. Patterns of invasive growth in vitro. Human decidua graviditatis confronted with established human cell lines and primary human explants. J Natl Cancer Inst. 1976;56:221-37. PMID:943555
- Prencipe M, Fitzpatrick P, Gorman S, Tosetto M, Klinger R, Furlong F, Harrison M, O'Connor D, Roninson IB, O'Sullivan J, et al. Cellular senescence induced by aberrant MAD2 levels impacts on paclitaxel responsiveness in vitro. Br J Cancer. 2009;101:1900-8. PMID:19935801
- Hirota Y, Cha J, Yoshie M, Daikoku T, Dey SK. Heightened uterine mammalian target of rapamycin complex 1 (mTORC1) signaling provokes preterm birth in mice. Proc Natl Acad Sci U S A. 2011;108:18073-8.
- Cha J, Bartos A, Egashira M, Haraguchi H, Saito-Fujita T, Leishman E, Bradshaw H, Dey SK, Hirota Y. Combinatory approaches prevent preterm birth profoundly exacerbated by gene-environment interactions. J Clin Invest. 2013;123:4063-75. PMID:23979163
- Deng W, Cha J, Yuan J, Haraguchi H, Bartos A, Leishman E, Viollet B, Bradshaw HB, Hirota Y, Dey SK. p53 coordinates decidual sestrin 2/AMPK/mTORC1 signaling to govern parturition timing. J Clin Invest. 2016;126:2941-54. PMID:27454290
- Burnum KE, Hirota Y, Baker ES, Yoshie M, Ibrahim YM, Monroe ME, Anderson GA, Smith RD, Daikoku T, Dey SK. Uterine deletion of Trp53 compromises antioxidant responses in the mouse decidua. Endocrinology. 2012; 153:4568-79. PMID:22759378
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A. 2007;104:16633-8. PMID:17921246
- Schmid P, Lorenz A, Hameister H, Montenarh M. Expression of p53 during mouse embryogenesis. Development. 1991;113:857-65. PMID:1821855
- Finn CA. The initiation of the decidual cell reaction in the uterus of the aged mouse. J Reprod Fertil. 1966;11:423-8. PMID:5940539
- Holinka CF, Finch CE. Age-related changes in the decidual response of the C57BL/6J mouse uterus. Biol Reprod. 1977;16:385-93. PMID:843564
- Goodrick GJ, Nelson JF. The decidual cell response in aging C57BL/6J mice is potentiated by long-term ovariectomy and chronic food restriction. J Gerontol. 1989;44:B67-71. PMID:2715583
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451-60. PMID:18692468
- Leontieva OV, Novototskaya LR, Paszkiewicz GM, Komarova EA, Gudkov AV, Blagosklonny MV. Dysregulation of the mTOR pathway in p53-deficient mice. Cancer Biol Ther. 2013;14:1182-8. PMID:24184801
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660-4. PMID:20457898
- Anders AP, Gaddy JA, Doster RS, Aronoff DM. Current concepts in maternal-fetal immunology: Recognition and response to microbial pathogens by decidual stromal cells. Am J Reprod Immunol. 2017; 77:e12623. PMID:28044385
- Gao L, Rabbitt EH, Condon JC, Renthal NE, Johnston JM, Mitsche MA, Chambon P, Xu J, O'Malley BW, Mendelson CR. Steroid receptor coactivators 1 and 2 mediate fetal-to-maternal signaling that initiates parturition. J Clin Invest. 2015;125:2808-24. PMID:26098214
- Lannon SM, Vanderhoeven JP, Eschenbach DA, Gravett MG, Adams Waldorf KM. Synergy and interactions among biological pathways leading to preterm premature rupture of membranes. Reprod Sci. 2014;21:1215-27. PMID:24840939
- Bonney EA, Krebs K, Saade G, Kechichian T, Trivedi J, Huaizhi Y, Menon R. Differential senescence in feto-maternal tissues during mouse pregnancy. Placenta. 2016;43:26-34. PMID:27324096
- Menon R, Boldogh I, Hawkins HK, Woodson M, Polettini J, Syed TA, Fortunato SJ, Saade GR, Papaconstantinou J, Taylor RN. Histological evidence of oxidative stress and premature senescence in preterm premature rupture of the human fetal membranes recapitulated in vitro. Am J Pathol. 2014;184:1740-51. PMID:24832021
- Menon R, Bonney EA, Condon J, Mesiano S, Taylor RN. Novel concepts on pregnancy clocks and alarms: redundancy and synergy in human parturition. Hum Reprod Update. 2016;22:535-60. PMID:27363410
- Menon R, Boldogh I, Urrabaz-Garza R, Polettini J, Syed TA, Saade GR, Papaconstantinou J, Taylor RN. Senescence of primary amniotic cells via oxidative DNA damage. PLoS One. 2013;8:e83416. PMID:24386195
- Menon R, Behnia, F, Polettini J, Saade G, Campisi J, Velarde M. Placenta membrane aging and HMGB1 signaling associated with human parturition. Aging. 2016;8:216-29. PMID:26851389
- Menon R. Human fetal membranes at term: Dead tissue or signalers of parturition? Placenta. 2016;44:1-5. PMID:27452431
- Leung TN, Zhang J, Lau TK, Hjelm NM, Lo YM. Maternal plasma fetal DNA as a marker for preterm labour. Lancet. 1998;352:1904-5. PMID:9863792
- Cha J, Bartos A, Egashira M, Haraguchi H, Saito-Fujita T, Leishman E, Bradshaw H, Dey SK, Hirota Y. Combinatory approaches prevent preterm birth profoundly exacerbated by gene-environment interactions. J Clin Invest. 2013;123:4063-75. PMID:23979163
- Cox LS, Redman C. The role of cellular senescence in ageing of the placenta. Placenta. 2017;52:139-145. PMID:28131318
- Cha J, Hirota Y, Dey SK. Sensing senescence in preterm birth. Cell Cycle. 2012;11:205-6. PMID:22189716