
ABSTRACT
Necroptosis is a form of programmed necrotic cell death mediated by the kinase RIPK3 and its substrate MLKL. MLKL, which displays plasma membrane (PM) pore-forming activity upon phosphorylation, functions as the executioner during necroptosis. Thus, it was previously assumed that MLKL phosphorylation is the endpoint of the necroptotic signaling pathway. Here, we summarize several events that characterize the dying necroptotic cells after MLKL phosphorylation, including Ca2+ influx, phosphatidylserine (PS) externalization, PM repair by ESCRT-III activation, and the final compromise of PM integrity. These processes add several unexpected regulatory events downstream of MLKL signaling. We have also observed that CoCl2, which may mimic hypoxia, can induce necroptosis, which suggests that in vivo triggers of necroptosis might include a transient lack of O2.
As a form of programmed cell death, necroptosis is regulated by the Ser/Thr kinase RIPK3 and its downstream substrate MLKL.Citation1 After MLKL is phosphorylated by RIPK3, MLKL undergoes a conformational change to release the N-terminal domain from auto-inhibition by the C-terminal pseudo-kinase domain, oligomerizes, and then binds to lipids in the plasma membrane (PM) or, possibly, the Golgi apparatus and endo-membrane systems.Citation2 The N-terminal domain, especially the four-helix bundle (4HBD), is sufficient to perforate membrane structures and execute cell lysis.Citation3 Given the direct pore-forming activity of p-MLKL, MLKL phosphorylation is often considered the final step of the necroptotic signaling cascade; however, we have demonstrated that cells with active MLKL are not always committed to death and can be resuscitated under certain conditions (see below), which suggests the existence of additional regulators.Citation4 Here, we summarize the biological processes and possible regulatory mechanisms downstream of MLKL activation and present new data to support our original observations.Citation4
Ca2+ influx during necroptosis requires MLKL activation
We recently reported Ca2+ influx during necroptosis that occurs immediately upon MLKL oligomerization.Citation4 In these experiments, oligomerization was triggered by a dimerization reagent, B/B, that binds to the tandem FKBP domains fused at the C-terminal of MLKL N-ter domain (1-181aa). We confirmed this Ca2+ influx by flow cytometry using a Ca2+ sensor protein, GCaMP3 (). MLKL activated by RIPK3-catalyzed phosphorylation also produced similar changes in intracellular Ca2+ levels (). To determine if the Ca2+ utilized for immediate influx is obtained from the extracellular culture media, we activated RIPK3 in Ca2+ free media. Under these conditions we observed a delay in MLKL-mediated Ca2+ flux (). Interestingly, intracellular Ca2+ ultimately increased before PM breakdown (). As there was no Ca2+ source in the culture media, we suggest that the Ca2+ was obtained from an intracellular Ca2+ pool, such as ER,Citation5 which may be damaged by active MLKL.Citation6 Because the MLKL1-181 truncation has no ability to bind to RIPK3, and because the NIH3T3 cells we used do not express RIPK3, we concluded that this Ca2+ influx does not involve RIPK3 activation.Citation4 In contrast, as MLKL silencing abolished Ca2+ influx in response to RIPK3 activation, we indicated that necroptotic Ca2+ influx requires MLKL.Citation4
Figure 1. Ca2+ influx occurs following MLKL activation and prior to plasma membrane breakdown during necroptosis. (A-C) Flow cytometric analysis of 100 nM B/B dimerizer-treated GCaMP3 expressing hMLKL1-181-2Fv-NIH3T3 cells (A), RIPK3-2Fv-NIH3T3 cells (B) and 20 ng/mL TNFα plus 100 μM zVAD (TZ) treated RIPK3-2Fv-NIH3T3 cells (C) stained with propidium iodide (PI) at the indicated time points. Cells were stimulated in DMEM (serum-free) media with or without Ca2+.
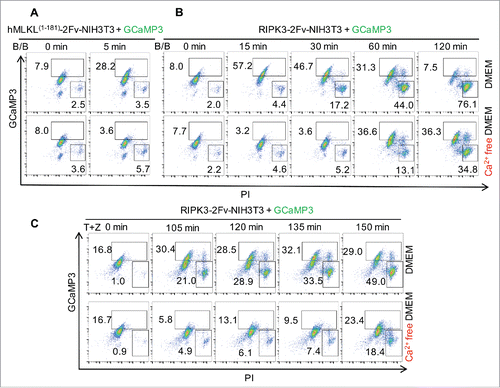
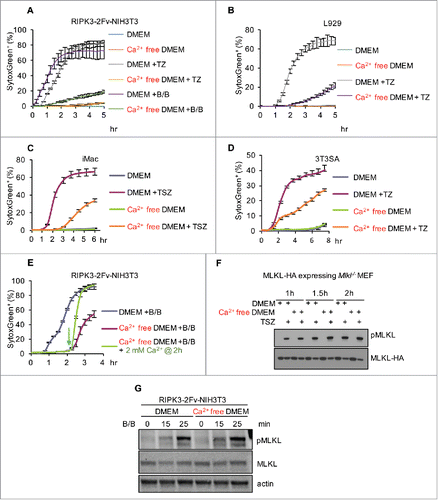
A requirement for Ca2+ influx in necroptosis remains controversial. For some cell lines, TRPM7-mediated Ca2+ influx (also requiring MLKL activation) was necessary for efficient cell lysis.Citation7 Other studies have also observed an ability of the Ca2+ chelator BAPTA-AM to delay the loss of PM integrity during necroptosis.Citation5,Citation7-10 On the other hand, it has been reported for different cell lines that it is not Ca2+, but rather other cations such as Mg2+ or Na+, that may be involved in necroptotic cell death.Citation6,Citation11,Citation12 In the cell lines we employed, including RIPK3-2Fv-NIH3T3, NIH3T3SA, immortalized macrophages (iMac), and L929 cells, extracellular Ca2+ influenced the progress to the final cell death (PI or SytoxGreen labeling, and ). Other studies found that while necroptosis could be delayed by the absence of Ca2+, the ultimate level of cell death was unaffected.Citation9 It is intriguing that cell lysis was tightly associated with intracellular Ca2+ abundance (), as lysis did not occur until the Ca2+ reached an apparently requisite level. Addition of 2 mM Ca2+ (close to the Ca2+ concentration in DMEM) to Ca2+ free media 2 hr after MLKL activation dramatically increased the rate of cell death as assessed by the PM-impermeable dye SytoxGreen (), which suggests that in the absence of Ca2+, necroptotic cells were trapped in a “primed” state without PM breakdown. We previously found that cells in Ca2+ free media did not display a defect in MLKL activation, as MLKL triggered the same extent of phosphatidylserine (PS) externalization, as discussed below.Citation4 Consistent with this idea, we observed that MLKL is phosphorylated independently of extracellular Ca2+ (). Therefore, while MLKL is activated and manifests its functions (e.g., PS exposure, see below) in the absence of elevated intracellular Ca2+, it appears that Ca2+ may be important for the dismantling of the PM by MLKL.
Figure 2. Ca2+ influx from the extracellular environment facilitates plasma membrane breakdown during necroptosis.(A-D) Incucyte quantification of SytoxGreen+ cells treated with 100 nM B/B, TZ or TSZ (20 ng/mL TNFα, 10 μM Lcl-161 plus 100 μM zVAD) as indicated in DMEM (serum-free) with or without Ca2+. (E) Incucyte quantification of SytoxGreen+ RIPK3-2Fv-NIH3T3 cells treated with 100 nM B/B. 2 mM Ca2+ was added at 2 hr post B/B stimulation. For all Incucyte quantification, data are presented as mean ± S.D. of at least triplicate samples. (F) Assessment of p-MLKL and MLKL (anti-HA) in MLKL-HA-MEF(Mlkl−/−) cells treated with TSZ in DMEM with or without Ca2+ for the indicated times. (G) Assessment of p-MLKL and MLKL (anti-MLKL) in RIPK3-2Fv-NIH3T3 cells treated with 100 nM B/B dimerizer in DMEM with or without Ca2+ for the indicated times.
PS externalization follows, but does not require Ca2+ influx
PS, a phospholipid, is distributed to the inner leaflet of the plasma membrane. During apoptosis, caspases induce a loss of PM asymmetry by several mechanisms, including activation of a scramblase and inhibition of a fllipase, resulting in exposure of PS on the cell surfaceCitation13-15. Consequently, PS externalization, which can be labeled by Annexin-V or MFG-E8, is widely considered a hallmark of apoptotic cells. However, we have found that phospholipid scrambling also occurs during necroptosis (specifically, after the Ca2+ influx and prior to plasma membrane breakdown).Citation4 The exposure of PS following activation of MLKL was also observed by others.Citation16 These findings clearly demonstrate that dying Annexin-V+; PI− cells may not necessarily be apoptotic. Another apoptotic event, caspase-3 activation, can also lead to Gasdermin E (DFNA5) cleavage, which induces pyroptosis-like secondary necrosis,Citation17,Citation18 and thus necrotic morphology does not preclude apoptosis. Going forward, the use of these “markers” to classify cell death must be used with care.
As mentioned above, although PS exposure occurs after Ca2+ influx, this process does not require the presence of extracellular Ca2+.Citation4 We further confirmed this using MFG-E8 (PS indicator) and X-rhod-1 (Ca2+ sensor). As shown in , cells in Ca2+ free media can scramble PS to the same extent with or without Ca2+ influx during necroptosis. Further, we found that the Ca2+-activated PS scramblase TMEM16F is not involved in PS externalization during necroptosis.Citation4 Moreover, in NIH3T3 cells, Ca2+ influx induced by the Ca2+ ionophores, ionomycin and A23187, failed to induce PS exposure despite elevating intracellular Ca2+, as indicated by GCaMP3 (). It is important to note that the key molecules responsible for PS exposure during necroptosis have not yet been identified. Because PS externalization can occur within 5 min of MLKL activation (), we speculate that MLKL action at the PM may directly contribute to PS externalization.
Figure 3. PS externalization occurs following Ca2+ influx and prior to plasma membrane breakdown during necroptosis. (A) Flow cytometric analysis of 200 nM B/B dimerizer-treated RIPK3-2Fv-NIH3T3 cells stained with MFG-E8-FITC at the indicated time points. Cells were preloaded with 2 μM X-rhod-1, AM for 30 min. (B) Flow cytometric analysis of 100 nM B/B dimerizer-treated GCaMP3-expressing hMLKL1-181-2Fv-NIH3T3 cells stained with Annexin V-APC at the indicated time points. Alternatively, cells were treated with 5 μM A23187 or 1 μg/mL Ionomycin. (C) Time lapsed confocal images of engulfment by peritoneal macrophages of TSZ treated (5.5 hr) Jurkat cells (necroptotic) expressing mCherry. Scale bar = 10 μm. (D) Flow cytometric quantification of the engulfment by peritoneal macrophages of apoptotic (20 ng/mL TNFα plus 80mJ/cm2 UV for 6 hr) and necroptotic (as in B) mCherry-expressing Jurkat cells. Cells with both mCherry and the macrophage marker CD11b-APC were considered to represent intact, engulfed cells. (E) Quantification of Annexn V-AF488 and TOPRO-3 fluorescence intensity over time by time lapsed confocal microscopy. HT-29 cells were treated with TSZ. (F) Time lapsed confocal images of hMLKL1-181-2Fv-NIH3T3 treated with 100 nM B/B and stained with Annexn V-AF488 and TOPRO-3. Scale bar = 10 μm.
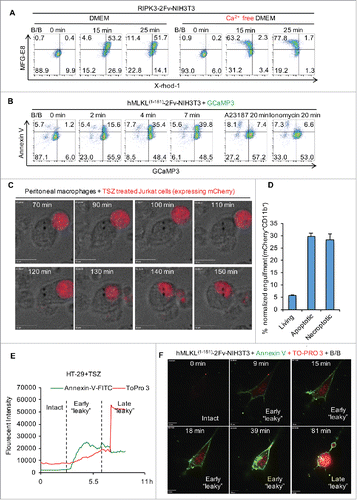
Dying cells expose PS and other “eat-me” signals to the surface, which can induce phagocytosis by dendritic cells and macrophages, a process called “efferocytosis”.Citation19 We observed that both apoptotic and necroptotic Jurkat cells were engulfed by neighboring peritoneal macrophages before loss of plasma membrane integrity. Using cells expressing mCherry, necroptosis was induced by treatment with TNF, Smac-mimetic, and zVAD-fmk (TSZ) and co-cultured with peritoneal macrophages. We observed engulfed cells that sustained PM integrity, as indicated by mCherry (). Thus, necroptotic PS re-distribution mechanisms may impact the clearance of cells dying by necroptosis, and thus may affect inflammation (e.g.,Citation20). Similar findings on phagocytosis of necroptotic cells has been reported by others as well.Citation16
Using live cell imaging, we also found that the PM of cells undergoing necroptosis may be slightly compromised, as evidenced by their becoming “leaky” to the dye TO-PRO-3 (MW = 671.42) before the collapse of the PM (). This early “leaky” phase during necroptosis suggests a counteracting damage-repair balance may occur during the course of necroptosis (discussed below). Cells that ultimately die when their self-repair activity is overwhelmed by MLKL pore-forming activity display a more dramatic TO-PRO-3 signal elevation (late “leaky” phase) compared to the early “leaky” phase (). The early “leaky” cells are most likely impermeable to protein-sized molecules (such as Annexin-V or MFG-E8) as these cells exclude 10 kDa dextran (). During apoptosis, cells similarly display a “leaky” phase for TO-PRO-3, due to the caspase-dependent activation of the Pannexin-1 channel.Citation21 Whether Pannexin-1 is involved in the MLKL-dependent necroptotic “leaky” phase is not known.
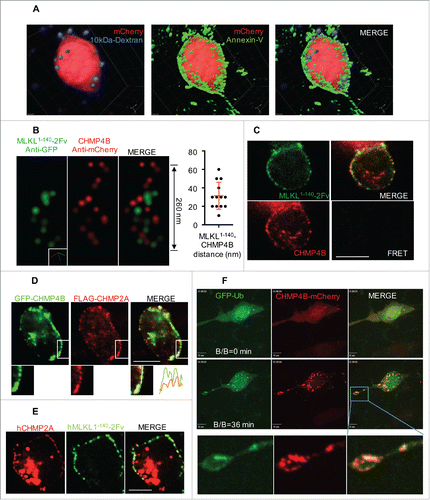
Figure 4. Plasma membrane shedding is mediated by ESCRT machinery during necroptosis. (A) Z-stack confocal microscope images of RIPK3-2Fv-NIH3T3 cells, treated with 100 nM B/B. Cells expressed mCherry (red) and were stained with AnnV-AF488 (green) and 10 kDa Dextran-NH2-AF647 (violet). (B) STORM images of the colocalization of CHMP4B and MLKL1-140-2Fv. Dox-inducible hMLKL(1-140)-2Fv-Venus in mlkl−/− MEF, stably expressed with hCHMP4B-mCherry, were treated with 2 μg/mL Dox overnight, followed by 25 nM B/B for 15 min. STORM was performed after anti-GFP and anti-mCherry immunostaining. The distance between CHMP4B and MLKL signals is shown on the right. (C) Confocal microscope images and FRET analysis of the cells in (B). Scale bar = 10 μm. (D) Confocal microscope images of RIPK3-2Fv-NIH3T3 cells, stably expressing flag-CHMP2A and transiently transfected with GFP-CHMP4B, treated with 100 nM B/B for 30 min. Scale bar = 10 μm. Lower panel shows the enlargement of indicated box, as well as the fluorescent intensities along the dashed line. (E) Confocal microscope images of Dox-inducible hMLKL(1-140)-2Fv-Venus in mlkl−/− MEF, stably expressing mCherry-flag-hCHMP2A, treated with 2 μg/mL Dox overnight, followed by 25 nM B/B for 15 min. Scale bar = 10 μm. (F) Time lapsed confocal images of RIPK3-2Fv-NIH3T3 cells, stably expressing hCHMP4B-mCherry and transiently transfected with GFP-Ubiquitin (Ub), treated with 100 nM B/B. Scale bar = 10 μm. Lowest panel shows the enlargement of indicated box.
Necroptotic cells shed MLKL-damaged plasma membrane until cell lysis
Z-stacked live images revealed that Annexin-V (or MGF-E8) labeled “bubbles” are released from the otherwise intact cells during necroptosis (). These bubbles are largely broken, as they do not contain cytosolic proteins (e.g., mCherry) and are permeable to 10 kDa dextran (). These membrane dynamics were also observed by transmission electron microscope (TEM).Citation4 A similar membrane shedding was observed by another group using ultracentrifugation, flow cytometry, and scanning electron microscopy (SEM).Citation16 In contrast to PS externalization, the PM membrane shedding was also prevented by depletion of extracellular Ca2+.
Notably, we found that these bubbles occur at the sites where the MLKL N-terminal domain targets the PM and that bubble formation requires the presence of MLKL.Citation4 We suggest that this membrane remodeling mechanism is a cell survival strategy, by which dying cells can release broken membranes and sustain PM integrity as a result. As we discuss below, cells lacking this mechanism more rapidly progress to cell lysis.
ESCRT-III components are responsible for broken membrane shedding
The necroptotic broken membrane release is mediated by the action of ESCRT-III components.Citation4 One of these components, CHMP4B, can translocate from the cytosol to sites where the bubbles occur, co-localizing with the MLKL N-terminal domain.Citation4 Using STORM, we examined this colocalization (). The average distance from CHMP4B to MLKL was approximately 20–30 nm (), which suggests that these proteins do not directly interact. Consistent with this finding, no fluorescence resonance energy transfer (FRET) was observed between CHMP4B and MLKL (). Active CHMP4B has been described to form a helical structure,Citation22 perhaps consistent with the STORM image we obtained ()
Another ESCRT III component, CHMP2A, similarly colocalized with both CHMP4B and MLKL1-140 during necroptosis (). This colocalization was more obvious upon loss of PM integrity, due to decreased background of cytosolic CHMP2A, leaving the PM-bound CHMP2A fraction. Silencing of CHMP2A or CHMP4B eliminated the formation and release of the bubbles from both human and murine cells.Citation4 Furthermore, using siRNA, we identified several other ESCRT components involved in this repair mechanism during necroptosis, including VPS4, IST1, and the ESCRT-I component TSG101.Citation4 As TSG101 has a ubiquitin binding domain, it was interesting that we observed colocalization of Ub and CHMP4B in some cells ().
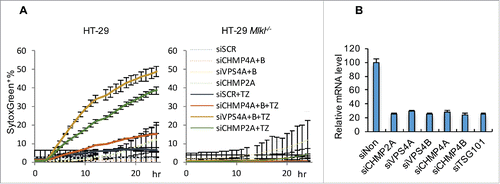
We previously provided evidence that silencing CHMP2A, CHMP4B, VPS4, IST1 or TSG101 can accelerate necroptosis, but not apoptosis, in murine cells (e.g. L929 and NIH3T3).Citation4 To determine if ESCRT can similarly counterbalance necroptotic stimuli in humans, we examined HT-29 cells, a human colorectal adenocarcinoma cell line (). As expected, HT-29 cells did not undergo necroptosis in response to TNF plus zVAD treatment (as Smac-mimetic is usually requiredCitation23), but we found that silencing either CHMP2A or VPS4A/B promotes MLKL-dependent cell death by this treatment (). Although CHMP4A and B were also largely silenced (75%), we failed to confirm a role of these two molecules (). It is possible that residual CHMP4A/B was sufficient for membrane repair in this setting.
Figure 5. ESCRT-III machinery antagonizes HT-29 necroptotic cell death. (A) Incucyte quantification of SytoxGreen+ HT-29 (left) and HT-29 Mlkl−/− (right) cells transfected with the indicated siRNA and stimulated with TZ (50 ng/mL TNF and 100 μM zVAD) over time. For all Incucyte quantifications, data are mean ± S.D. of at least triplicate samples. (B) Efficiency of siRNA silencing in HT-29 cells, determined by qRT-PCR.
Others have reported that PM damage by various treatments, including laser, digitonin, and bacterial toxins, can be repaired by ESCRT.Citation24,Citation25 Here, we provide another example. During necroptosis, MLKL pore-forming activity can be neutralized by ESCRT-mediated membrane repair and membrane shedding. Ca2+ has been shown to be required for ESCRT-mediated plasma membrane repair,Citation24,Citation25 and, consistent with this, we observed that depletion of extracellular Ca2+ prevented CHMP4B membrane translocation and the formation of PM bubbles.Citation4
It seems contradictory that although Ca2+ appears to be required for ESCRT-mediated PM repair, cells tend to die more slowly in the absence of extracellular Ca2+. One possible explanation is that Ca2+ controls both MLKL-triggered pore-forming and repair processes, with the first upstream of the second. To fully address this puzzle, the Ca2+ sensors for each process must be identified. Although the accessory proteins ALG2 and Alix were implicated in recognition of Ca2+ in response to laser damage,Citation24,Citation25 we were unable to identify roles for these proteins in the response to active MLKL by siRNA silencing.Citation4 There are only limited numbers (several hundred) of Ca2+ binding proteins encoded in the human genome, and therefore it may be feasible test these candidates by a well-designed small pool siRNA screen.
ESCRT maintains the survival of cells with active MLKL
As discussed above, phosphorylated MLKL (p-MLKL) can damage PM and thus lead to cell lysis. Consequently, phosphorylation of MLKL is thought to be the last and irreversible step of necroptosis and is often used as a marker of necroptosis. However, we and others have demonstrated that cells with p-MLKL may not be committed to cell death if the p-MLKL is inactivated before cell lysis (e.g., via addition of the MLKL inhibitor necrosulfonamide or inactivation of RIPK3).Citation4,Citation16 Provided the actions of ESCRT machinery sustain cell survival, the cells can regain PS asymmetry, lower intracellular Ca2+ levels, and inactivate MLKL. This phenomenon, termed “resuscitation”, may also happen in vivo.Citation4 Biopsies from human kidneys shortly after transplantation showed apparently healthy endothelial cells that were positive for p-MLKL staining, and this was associated with increased expression of several ESCRT components. Elevation of ESCRT components was also observed in murine kidney following ischemia-reperfusion injury. Based on these findings, we might predict the existence of signals that can coordinately induce the expression of ESCRT.Citation4 The elucidation of such processes could have important clinical applications; among other promising lines of inquiry, it will be interesting to determine if ESCRT expression predicts the outcome of organ transplantation.
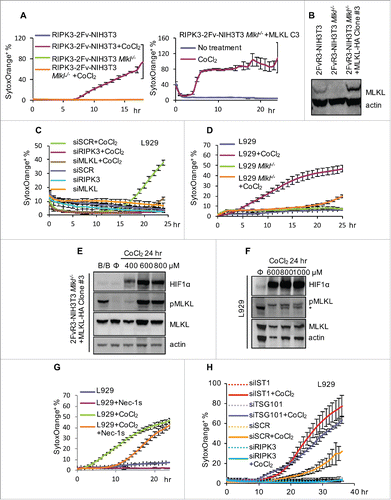
Recently, several in vitro studies and animal models have suggested that hypoxia can activate RIPK1, RIPK3, and perhaps MLKLCitation26-30. Hypoxia induces HIF-1 stabilization by inhibition of prolyl hydroxylase (PHD), and we examined the effects of CoCl2, which can inhibit PHD activity. We found that CoCl2 treatment strongly induced RIPK3- and MLKL-dependent cell death in both NIH3T3 and L929 cells (). We also observed the induction of HIF-1α and the accumulation and MLKL phosphorylation upon treatment (). Given that no Caspase-8 inhibitor was added to these cell cultures, it is intriguing that this hypoxia-mimetic can drive the machinery of necroptosis. RIPK1 appeared to be less involved in this process, as Nec-1s only slightly attenuated cell death (). The silencing of ESCRT components TSG101 and IST1 accelerated this cell death (). It is unclear if hypoxia, per se, or some other effect of CoCl2 mediated necroptosis in this system. Nevertheless, these findings have important implications for studying heavy metal toxicity.
Figure 6. Hypoxia mimicking reagent CoCl2 induces necroptosis. (A) Incucyte quantification of SytoxOrange+ RIPK3-2Fv-NIH3T3 cells (WT, Mlkl−/−, or Mlkl−/− reconstituted with mMLKL-HA clone #3) treated with 600 μM CoCl2. (B) Verification of MLKL expression in cells used in (A). Note that clone #3 had a higher level of MLKL than WT, which may explain why clone #3 died more rapidly than WT cells. (C-D) Incucyte quantification of SytoxOrange+ L929 cells (WT or Mlkl−/−, or transfected with indicated siRNA) treated with 800 μM CoCl2. (E-F) Assessment of p-MLKL and HIF1α in CoCl2-treated (24 hr) RIPK3-2Fv-NIH3T3 clone #3 (E) and L929 (F) cells, determined by western blot. * indicated non-specific band. (G) Incucyte quantification of SytoxOrange+ L929 cells (with or without 60 μM Nec-1s) treated with 800 μM CoCl2. (H) Incucyte quantification of SytoxOrange+ L929 cells (with indicated siRNA) treated with 800 μM CoCl2. For all Incucyte quantification, data are mean ± S.D. of at least triplicate samples.
Other events following MLKL activation
After MLKL activation, a small portion of MLKL is translocated to the nucleus.Citation31 Although the physiological significance of this translocation remains unclear, it is possible that nuclear MLKL can change the transcriptome to promote inflammation. It is also suggested that MLKL nuclear translocation might facilitate cell death, in that the MLKL N-terminal truncation, which loses the capability to target the nucleus, correspondingly has less activity to induce cell lysis,Citation31 consistent with what we have reported.Citation4
Before the integrity of the PM is compromised, evidence suggests that active MLKL can also lead to the cleavage and general shedding of several cell-surface proteins (e.g., ectodomain) processed by cell-surface proteases of the “a disintegrin and metalloprotease” (ADAM) family.Citation32 After MLKL is phosphorylated, it can form a complex with ADAMs, which together facilitate cell death, inflammation, cell migration, and invasion. As more than 100 ADAM substrates have been identified thus far, these proteins may function as signal messengers to extend the biological consequences of necroptosis.
MLKL activation also leads to the transcription and expression of chemokines (such as CXCL1 and CXCL10) in a RIPK3-independent manner, an effect that is observed even when only the N-terminal domain of MLKL is dimerized.Citation4 As this induces a Ca2+ influx, it is possible that the intracellular Ca2+ signal participates in this induction.
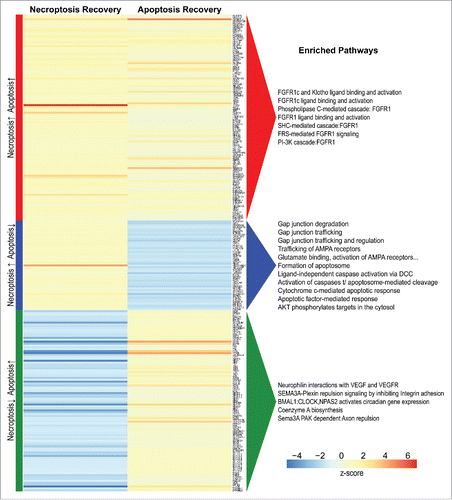
As in the process of necroptosis, it appears that cells that engage caspase activation by stimuli that promote apoptosis can also survive if the stimulus is withdrawn.Citation33 This process, called “anastasis” has parallels with resuscitation of cells undergoing necroptosis. Changes in gene expression during anastasis have been reported.Citation34 We propose that comparisons between necroptosis-resuscitation and apoptosis-resuscitation might allow us to identify novel markers for these conditions. Utilizing expression data from cells that survive either process, we identified a set of genes that were significantly upregulated in both recovery conditions as well as two sets of genes that were uniquely regulated in each type of resuscitation (, Table S1). A “resuscitation signature” for each type of cell death may exist within these candidates, perhaps as a suite of multiple genes enriched in the same pathway with moderate differences in expression.
Figure 7. Genes upregulated during the recovery of apoptosis or necroptosis. Heatmap showing differential gene expression between resuscitated and control samples from necroptosis and apoptosis experiments, with log2-fold change (LFC) of average expression scaled to z-score (columns). Data on apoptosis resuscitation (anastasis) were kindly provided by Dr. Denise Monach (UC Santa Barbara)Citation34. Differential expression was modeled separately for each experiment, and significant genes (FDR < 0.05) with an LFC ≥ 0.5 in both experiments were grouped according to concordance/discordance of signal between the experiments. Enriched pathways (FDR < 0.05) for each visualized group were obtained from Reactome gene set analysis.
Discussion
Immediately following activation of MLKL, Ca2+ influx from the extracellular environment occurs. Following but independent of this event, PS is scrambled to the outer leaflet of the PM. Then, driven by the increase in intracellular Ca2+, ESCRT components accumulate at the MLKL-damaged PM sites and facilitate membrane repair by releasing the damaged membrane domains as bubbles. At this stage, if the necroptotic stimulus can be removed or blocked, the dying cells can be resuscitated and survive. In contrast, if the necroptotic stimulus continues, the pore-forming activity of activated MLKL overcomes the ESCRT repair activity, thereby committing the cell to lysis.
ESCRT-mediated plasma membrane repair can also prevent necroptosis. The silencing of ESCRT can sensitize HT-29 cells to necroptosis induced by TNF plus zVAD (). As this treatment fails to cause necroptosis in these cells when ESCRT is intact, these results strongly suggest that cells can survive limited levels of active MLKL. We observed a similar effect in ESCRT-silenced L929 cells, rendering them sensitive to necroptosis induced by TNF treatment alone.Citation4 It is important to note that while the membrane repair process engaged by ESCRT may be responsible for this effect, ESCRT may also have other roles in regulating the function of active MLKL at the PM.
Recently, Wallach and colleaguesCitation35 reported that MLKL can promote the release of MLKL containing small vesicles (∼150 nm) from cells via ESCRT activation, which was augmented by RIPK3. They also suggest that ESCRT activation during necroptosis can facilitate the lysosomal degradation of several membrane receptors as well as membrane-targeted p-MLKL. We, however, found no difference in p-MLKL with or without ESCRT,Citation4 but instead found that ESCRT acts downstream of MLKL activation. Both studies, however, found that ESCRT can promote cell survival and therefore allow cells to produce more cytokines and chemokines that may contribute to cellular communication.Citation4,Citation35,Citation36 They also found a physical association of MLKL and the ESCRT complex, using mass spectroscopy as well as western blot, further strengthening the molecular link between necroptosis and membrane remodeling.Citation35 Although we found no direct binding between MLKL and CHMP4B (), interaction with other ESCRT components would be consistent with the co-localizaiton we observed ().
These biological events during necroptosis and the molecular mechanisms behind them require further investigation. Is there a channel responsible for MLKL-induced Ca2+ influx? How is PS exposed during necroptosis? What does ESCRT sense in order to translocate to the broken sites punctured by MLKL? Is there a phosphatase that can remove the phosphate from p-MLKL? What is the role of Ca2+ in necroptosis? Although MLKL's role as an executioner molecule has been well characterized, we lack a comprehensive understanding of its functions.
A key question in the study of necroptosis is when and why necroptosis occurs in vivo. In studies investigating this question, p-MLKL is often used as a marker to label “necroptotic” cells in tissue samples. This approach suffers, however, from the lack of a murine p-MLKL antibody for histological studies, which hampers our ability to further confirm whether the p-MLKL staining pattern in human samples is truly specific using murine MLKL knockout samples. Furthermore, murine studies with genetically modified animals are potentially confounded by the roles that RIPK3 and MLKL play in other processes unrelated to necroptosis. Due to these limitations, the role of in vivo necroptosis in physiological or pathological conditions has not been well characterized thus far. Moreover, studies of necroptosis in vitro generally require a block of caspase-8, which may be an uncommon physiological setting.Citation37 However, our findings suggest a basal level of MLKL activation can occur without caspase-8 inhibition and counterbalanced by ESCRT-III,Citation4 which may further raise the possibility that p-MLKL need not necessarily lead to cell elimination in vivo.
Although necroptosis is a form of programmed cell death, the extent of the complete program's complexity is unknown. Despite these shortcomings, progress in this field has led to a better understanding of the pathology of numerous diseases; in some circumstances, such work has facilitated the identification of therapeutic targets and putative therapeutic compounds that have shown potential for clinical application.Citation1 The ability to rescue cells from programmed death, or even to predict whether certain cells are more or less poised to recover from necroptosis, would likewise have implications for patient outcomes.
Materials and methods
Compounds, antibodies and reagents
The dimerizer (B/B) was purchased from Clontech. The cytokines used were: mTNFα (Peprotech) and hTNFα (Peprotech). The SMAC mimetic and inhibitor of IAP (LCL-161) was purchased from Chemietek. Other reagents included 7-Cl-O-Nec-1 (Nec-1s, 504297, calbiochem), zVAD-fmk (A1902, Apexbio) and CoCl2 (Sigma). Antibodies used for immunoblotting in this study were anti-actin (sc-1616, Santa Cruz), anti-FLAG (A8592, Sigma), anti-HA (H6908, Sigma), anti-mMLKL (AP14272b, Agent), anti-pMLKL (ab187091, Abcam) and anti-HIF-1α (10006421).
Plasmids
Flag-hCHMP2A, “humanized” mMLKL-HA, truncated hMLKL with two Fv domains and HA tag were cloned into pMX-IRES-mCherry. Flag-MLKL was cloned into pMX-IRES-GFP. hCHMP4B-mCherry was cloned into pBabe-puro. GCaMP3 was cloned into pLZRS vector. GFP-Ub was cloned into pEGFP-C1. GFP-CHMP4B expressing plasmid was a gift from Dr. Paul Bieniasz (Rockefeller University). The hCHMP4B-mCherry plasmid was a gift from Dr. Jennifer Lippincott-Schwartz (Janelia Research Campus).
Cell lines
NIH3T3, L929, MEF and immortalized macrophages (iMacs) were maintained at 37°C, 5% v/v CO2 in a humidified incubator in DMEM (GIBCO) supplemented with 10% FBS, 2 mM L-glutamine (GIBCO), 200 U/mL penicillin-–streptomycin (GIBCO). HT-29 cells were cultured using the same conditions except McCoy's 5A media (GIBCO). Jurkat cells were cultured in RPMI-1640 media (GIBCO) with the same supplements as above. All media and supplements were purchased from Life Technology. All media were supplemented with 10 μg/mL plasmocin (Invivogen). The HT-29 and Jurkat cell lines are authenticated by the University of Arizona Genetics Core. Primary peritoneal macrophages were prepared following the standard protocols.Citation38
RIPK3-2Fv-NIH3T3 cells, hMLKL1-181-2Fv-NIH3T3, MLKL-HA-MEF(Mlkl−/−) and hMLKL1-140-2Fv-Venus-MEF(Mlkl−/−) were previously described.Citation4 GCaMP3, “humanized” MLKL-HA and CHMP4B-mCherry are stably expreesed by retroviral transduction. NIH3T3(Mlkl−/−), L929(Mlkl-/-) and HT-29(Mlkl−/−) cell lines were generated by CRSPR/Cas9 plasmid pSpCas9(BB)-2A-GFP (PX458) and has been described in our previous study.Citation4
siRNA
All of the “on-target” siRNA (smart-pool) were purchased from Dharmacon. siRNA transfection was performed by Lipofectamin RNAiMax (Life technology) on HT-29 and INTERFERin (polyplus transfection) on L929 cells, respectively. RNAi efficiency was assessed by realtime PCR at 60 h post transfection or cell death assay at 66–70 h post transfection.
Flow cytometric quantification of cell death
To assess necroptosis, cells were trypsinized, resuspended in serum-free DMEM and stained with MGF-E8-FITC (1:50, Haematologic Technologies), Annexin V-APC (1:50-100, eBioscience), 50 nM SytoxGreen (invitrogen) or 2 μg/ml PI (Sigma) for 5 min. For X-rhod-1, AM preloading, cells were cultured in complete media with 2 μM X-rhod-1, AM for 30 min at 37°C, 5% v/v CO2. Before cell death stimulation, cells were washed once by HBSS. Data were analyzed by flow cytometry using FACsCalibur systems (BD Biosciences) and FlowJo Collectors' Edition software (Tree Star). The percentages of differently labeled cells were calculated by FlowJo software (Tree Star).
Real-time PCR
Total RNA for real-time PCR was prepared using the RNeasy Mini Kit (Qiagen). Reverse transcription reactions were catalyzed by M-MLV reverse transcriptase (Life Technology), following the standard protocol using random hexamers (IDT). Real-time PCR was performed with SYBR™ Green labeling in 7500 Fast Real-Time PCR System (Applied Biosystems). PCR conditions were 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 15 s and 60°C for 1 min. mRNA expression was normalized against actin, therefore allowing comparison of mRNA levels. Primers used for qPCR are CATGTACGTTGCTATCCAGGC, CATGTACGTTGCTATCCAGGC (actin); CGCGAGCGACAGAAACTAGAG, CCCGCATCAATACAAACTTGC (hCHMP2A); TCAGCGTGAGGCCATTGAG, GTTGTTCCGTGATGTCAGTCA (hCHMP4A); TGCAGAGGAGATTTCAACAGC, TGTTTCGGGTCCACTGATTTC (hCHMP4B); ATGGCTACTGGACACATACCC, GCGGATAGGATGCCGAAATAG (hTSG101); GTGATGGAGAAGCCCAACATAC, CAAGTGTGGGAATTTGATTGGC (hVPS4A); and CGACCAAATGTGAAATGGAGTGA, TCCAGGCGGCCCAAATAATAG (hVPS4B).
IncuCyte analysis
For IncuCyte analysis, cells were first seeded into 12 or 24 well plates one day before the assays. On the other day, 50 nM SytoxGreen (Molecular Probes) plus the indicated cell death stimuli were added, and then, cells were moved into an IncuCyte live cell imaging system (http://www.essenbioscience.com/en/products/incucyte/?gclid = CJ7ywJynytICFd63wAodCeEALA). Cells were imaged every 0.5 h or 1 h and the SytoxGreen labeled cells (counted as dead cells) were quantified by the IncuCyte FLR or Zoom software (http://www.essenbioscience.com/en/products/software/). When percentages are calculated, total cell number was quantified at the end of each course of treatment, using 200 μg/ml digitonin (Sigma-Aldrich) to permeabilize all cells that were stained with 50 nM SytoxGreen. Data were then shown as a percentage of SytoxGreen+ cells to total cell numbers. At least two experiments were performed with four replicates for each condition.
Alternatively, 50 nM SytoxOrange (Molecular Probes) plus 50 nM Syto16 (Molecular Probes) together with cell death stimuli were added. As SytoxOrange only labels dead cells while Syto16 can label all the cells, the percentage of SytoxOrange+ cells to total cell numbers can be calculated.
Confocal microscopy
Cells were plated on 2-well or 4-well glass chamber slides (Mattek) coated with fibronectin (100 µg/mL in PBS, Millipore), one day before analysis. Cells were maintained in complete media at 37°C and 5% CO2 in a climate controlled chamber (Solent Scientific, UK). Annexin V-Alexa Fluor-488 (Molecular Probes) or Annexin V-FITC (eBioscience) was added into the media to label PS externalization (1:100). As indicated, TOPRO-3 (500 nM, Molecular Probes) was added into media to label the nuclear. Cells were then stimulated with B/B dimerizer or TSZ before confocal microscopy analysis. To measure Dextran uptake, 10 kDa Dextran-NH2-Alexa Fluor 647 (10 μg/mL, Molecular Probes) and Annexin V-Alexa Fluor-488 (molecular probes) were added to the dying cells for 5 min, then without wash, immediately fixed with 4% paraformaldehyde (Electron Microscopy Science, diluted by serum free DMEM, with additional 5 mM CaCl2) for 15 min at room temperature. Then the cells were washed once with serum free DMEM and examined by confocal microscopy. For flag staining, flag antibody (M2, Sigma) was applied according to the product technical bulletin.
Confocal microscopy was performed with a Marianas spinning disk confocal imaging system (Intelligent Imaging Innovations/3i) consisting of a CSU-22 confocal head (Yokogowa Electric Corporation, Japan); solid-state diode-pumped laser launch (3i) with wavelengths of 445 nm, 473 nm, 523 nm, 561 nm, and 658 nm; and a Carl Zeiss Axiovert 200M motorized inverted microscope equipped with a precision motorized XY stage (Carl Zeiss MicroImaging) and spherical aberration correction optics (3i). The images were analyzed with Slidebook 6.
Super resolution fluorescence microscopy
Stochastic optical reconstruction microscopy (STORM) was performed as recently described.Citation39 Briefly, cells were fixed with 4% paraformaldehyde followed by treatment with sodium borohydride to quench free reactive groups. Cells were subsequently permeabilized for 3 min in buffer (50 mM Tris-HCl, pH8.0, 50 mM NaCl) containing 0.1% Triton-100 prior to blocking in buffer containing 2% BSA and 0.05% Tween-20. Samples were incubated overnight in TBS buffer containing BSA, anti-GFP (Biolegend, cat#338002) and anti-mCherry (Abcam, cat#167453) antibodies, and detected with CF568 and CF647 labeled secondary antibodies (Biotium). 3-dimensional STORM acquisition was facilitated using an N-STORM system (Nikon Instruments) comprised of a 100 × 1.49NA TIRF objective and an astigmatic lens inserted into the light path, before collection using an iXon DU897 ultra EMCCD camera with frame rate of 109 f.p.s. and EM gain of 17 mHz at 16 bit, as previously described.Citation39 Samples were imaged in buffer comprised of 50 mM Tris-HCL, 50 mM NaCl and 10% glucose, containing 10 mM cysteamine and oxygen scavengers. Images were processed using algorithms previously described Citation40 and incorporated into Elements imaging software (Nikon Instruments).
Western blotting
Cells were lysed directly with loading buffer (50 mM Tris pH = 6.8, 2%SDS and 10% glycerol) and denatured by heating. Protein concentration was determined by a BCA assay (Pierce) and systematically normalized before loading on SDS-PAGE. Following the transfer to Hybond C nitrocellulose (Amersham Bioscience), immunodetection was performed using the indicated primary and peroxidase-coupled secondary antibodies (GE). Proteins were visualized by enhanced chemiluminescence (ECL, GE).
For HIF1-α blotting, the antibody 10006421 (Cayman) was used. For p-MLKL detection in L929 and NIH3T3 cells, the antibody ab196436 (Abcam) was used. For p-MLKL detection in MEF cells, a construct expressing a “humanized” mouse MLKL was stably introduced. In this construct, the human MLKL sequence “ELRKTQTSMSLGTTR”, which is the RIPK3 phosphorylation site, replaced the mouse MLKL sequence “ELSKTQNSISRTAKS”. This “humanized” mouse MLKL is able to be phosphorylated by mouse RIPK3 and also recognized by human p-MLKL antibody ab187091 (Abcam).
In vitro phagocytosis assay
Apoptosis was induced in Jurkat cells expressing mCherry by treatment with TNFα (20 ng/mL) plus UV irradiation (80 mJ/cm2) for 6 hr. Necroptosis was induced in Jurkat cells expressing mCherry by treatment with TSZ for 5.5 hr. Before the phagocytosis assay, dying cells were analyzed by FACS to determine the percentage of Annexin-V+, SytoxGreen− cells, which were used for normalization later. Dying Jurkat cells were added to peritoneal macrophage cultures at a ratio of 1:3 (dead cell: macrophage). After spinning at 350 g for 5 min, the cells were back into incubator for 1 hr or examined by time-lapse confocal imaging. After incubation, macrophages and Jurkat cells were collected together and stained with CD11b-APC (eBioscience) for 10 min and assessed by flow cytometry. We calculated how many Jurkat cells could be engulfed by macrophages in each condition (mCherry+CD11b+/total mCherry+). For normalization, only Annexin-V+ SytoxGreen− cells were counted as total mCherry+ cells for apoptotic and necroptotic conditions.
Expression analyses
Necroptosis was induced by addition of B/B dimerizer to NIH3T3 cells expressing MLKL1-181-2Fv for 1 hr, AnnV+ cells were sorted, and then treated with “washout” (Clonetech) for 6 hr to cause resuscitation, and subjected to microarray analysis as describedCitation4 (Gene Expression Omnibus Accession GSE85660). Data from untreated control and resuscitated samples (n = 3 for each) were corrected for background noise, quantile normalized, and median-polish summarized in R using the RMA method,Citation41 as implemented in the BioConductor package oligo (v1.40.1).Citation42 Affymetrix probe set identifiers were annotated using the BioConductor package AnnotationDbi (v1.38.1)Citation43 with the mogene20sttranscriptcluster database (v8.6.0).Citation44 Differential expression between control and resuscitated samples was tested using per-gene linear models and an empirical Bayes estimation of expression variances, as implemented in the BioConductor package limma (v3.32.2).Citation45 P-values were adjusted for multiple testing by applying the Benjamini & Hochberg false discovery rate (FDR) method.
Differentially expressed genes from an RNA-Seq experiment studying apoptosis-resuscitation (anastasis) were kindly provided by Sun and colleagues for comparison to necroptosis-resuscitation.Citation34 For this comparison, we used the recovery time point most comparable to that of the necroptosis experiment (8 hr), again utilizing only those genes that were significantly differentially expressed (FDR < 0.05) between control and resuscitation conditions.
To compare expression between necroptosis- and apoptosis- resuscitation, we focused on the gene sets that were either upregulated in both resuscitation conditions (Necroptosis↑ Apoptosis↑) or upregulated in one condition and downregulated in the other (i.e, Necroptosis↑ Apoptosis↓; Necroptosis↓ Apoptosis↑). We then calculated a z-score of relative expression in each experiment by scaling the log2-fold change (LFC) values from all of these genes, regardless of gene set. The z-scores of genes that were differentially expressed at ≥ 0.5 LFC in both experiments were visualized using the R package NMF (v0.17.6)Citation46 with designated ordering of genes based on signal concordance between experiments. These same gene sets were also analyzed for Mus musculus pathway enrichment using the Reactome Pathway Knowledgebase with options “Project to human” and “Include interactors” both disabled.Citation47 FDR was again used to adjust for multiple comparisons.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCCY_A_1371889_Supplement.docx
Download MS Word (53.3 KB)Acknowledgments
We thank Dr. Denise Monach (UC Santa Barbara) for providing us RNAseq data of anastasis. We are grateful to all the Green lab members for helpful discussion, suggestions and collaborations.
Funding
This work was supported by grants from the US NIH (R01 CA169291 and R01 AI44828) and ALSAC.
References
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455-65. doi:10.1056/NEJMra1310050. PMID:24476434
- Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017. doi:10.1038/cdd.2017.65. PMID:28498367
- Zhang J, Yang Y, He W, Sun L. Necrosome core machinery: MLKL. Cell Mol Life Sci. 2016;73(11-12):2153-63. doi:10.1007/s00018-016-2190-5. PMID:27048809
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169(2):286-300 e16. doi:10.1016/j.cell.2017.03.020
- Ousingsawat J, Cabrita I, Wanitchakool P, Sirianant L, Krautwald S, Linkermann A, Schreiber R, Kunzelmann K. Ca2+ signals, cell membrane disintegration, and activation of TMEM16F during necroptosis. Cell Mol Life Sci. 2017. 74(1):173-181. doi:10.1007/s00018-016-2338-3. PMID:27535660
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133-46. doi:10.1016/j.molcel.2014.03.003
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55-65. doi:10.1038/ncb2883. PMID:24316671
- Sun W, Wu X, Gao H, Yu J, Zhao W, Lu JJ, Wang J, Du G, Chen X. Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Radic Biol Med. 2017. 108:433-444. doi:10.1016/j.freeradbiomed.2017.04.010
- Ros U, Peña-Blanco A, Hänggi K, Kunzendorf U, Krautwald S, Wong WW, García-Sáez AJ. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017;19(1):175-187. doi:10.1016/j.celrep.2017.03.024
- Nomura M., Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014;74(4):1056-66. doi:10.1158/0008-5472.CAN-13-1283. PMID:24371227
- Xia B., Fang S, Chen X, Hu H, Chen P, Wang H, Gao Z. MLKL forms cation channels. Cell Res. 2016;26(5):517-28. doi:10.1038/cr.2016.26. PMID:27033670
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, et al., Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105-21. doi:10.1038/cr.2013.171. PMID:24366341
- Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403-6. doi:10.1126/science.1236758. PMID:23845944
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164-8. doi:10.1126/science.1252809. PMID:24904167
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of `plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182(5):1545-56. PMID:7595224
- Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, Regev-Rudzki N, Edry-Botzer L, Gerlic M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15(6):e2002711. doi:10.1371/journal.pbio.2002711. PMID:28650960
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi:10.1038/ncomms14128. PMID:28045099
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a Gasdermin. Nature. 2017; 547(7661):99–103. doi:10.1038/nature22393. PMID 28459430
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396-401. doi:10.1073/pnas.1113421108. PMID:21969579
- Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, et al., Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115-9. doi:10.1038/nature17950. PMID:27096368
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, et al., Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467(7317):863-7. doi:10.1038/nature09413. PMID:20944749
- Mierzwa BE, Chiaruttini N, Redondo-Morata L, von Filseck JM, König J, Larios J, Poser I, Müller-Reichert T, Scheuring S, Roux A, et al., Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat Cell Biol. 2017;19(7):787-798. doi:10.1038/ncb3559. PMID:28604678
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100-11. doi:10.1016/j.cell.2009.05.021
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK. Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5:5646. doi:10.1038/ncomms6646. PMID:25534348
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136. doi:10.1126/science.1247136. PMID:24482116
- Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013;4:e622. doi:10.1038/cddis.2013.149. PMID:23640464
- Xu Y, Wang J, Song X, Qu L, Wei R, He F, Wang K, Luo B. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci Rep. 2016;6:29362. doi:10.1038/srep29362. PMID:27377128
- Vieira M, Fernandes J, Carreto L, Anuncibay-Soto B, Santos M, Han J, Fernández-López A, Duarte CB, Carvalho AL, Santos AE. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis. 2014;68:26-36. doi:10.1016/j.nbd.2014.04.002
- Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H1, Zhang J, et al., CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175-82. doi:10.1038/nm.4017. PMID:26726877
- Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13(11):2797-804. doi:10.1111/ajt.12448. PMID:24103029
- Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23(2):253-60. doi:10.1038/cdd.2015.92. PMID:26184911
- Cai Z, Zhang A, Choksi S, Li W, Li T, Zhang XM, Liu ZG. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26(8):886-900. doi:10.1038/cr.2016.87. PMID:27444869
- Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, Wong CS, Wu HY, Law HT, Liu K, et al., Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23(12):2240-52. doi:10.1091/mbc.E11-11-0926. PMID:22535522
- Sun G, Guzman E, Balasanyan V, Conner CM, Wong K, Zhou HR, Kosik KS, Montell DJ. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J Cell Biol. 2017; pii:jcb.201706134. doi:10.1083/jcb.201706134. PMID:28768686
- Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity. 2017;47(1):51-65 e7. doi:10.1016/j.immuni.2017.06.001
- Vandenabeele P, Riquet F, Cappe B. Necroptosis: (Last) Message in a Bubble. Immunity. 2017;47(1):1-3. doi:10.1016/j.immuni.2017.07.002
- Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56(4):469-80. doi:10.1016/j.molcel.2014.11.001
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008. Chapter 14:Unit 14 1. doi:10.1002/0471142735.im1401s83. PMID:19016445
- Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, et al. Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun. 2014;5:5645. doi:10.1038/ncomms6645. PMID:25487526
- Huang B, Wang W, Bates M, Zhuang X Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319(5864):810-3. doi:10.1126/science.1153529. PMID:18174397
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249-64. doi:10.1093/biostatistics/4.2.249
- Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363-7. doi:10.1093/bioinformatics/btq431. PMID:20688976
- Pagès, H., et al., AnnotationDbi: Annotation Database Interface. R package version 1.38.1. 2017.
- MacDonald, J., mogene20sttranscriptcluster.db: Affymetrix mogene20 annotation data (chip mogene20sttranscriptcluster). R package version 8.6.0. 2017.
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007. PMID:25605792
- Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. doi:10.1186/1471-2105-11-367. PMID:20598126
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, et al., The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44(D1):D481-7. doi:10.1093/nar/gkv1351. PMID:26656494