
p21 (CDKN1A, also known as p21WAF1/Cip1) is a 165 amino acid protein that can mediate both p53-dependent and p53-independent G1 growth arrest.Citation1 p21 executes its biological activities primarily by binding and inhibiting the kinase activity of the cyclin-dependent kinases (CDKs), in particular CDK4 and CDK6, thereby preventing phosphorylation of the retinoblastoma protein (pRB) and thus, in turn, suppressing the expression of proliferation-associated genes. This inhibition is one of the mechanisms which promotes a permanent form of cell cycle arrest, known as cellular senescence. Cellular senescence halts cell proliferation in response to various stressors, including telomere shortening, oncogene activation, cell-cell fusion of normal cells and DNA damage.Citation2 The senescence program facilitates protection from tumorigenesis and aids in tissue repair.Citation2 Conversely, accumulation of senescent cells in tissues, like the one which occurs in tissues with age, can contribute to age-related pathologies.Citation2 This deleterious accumulation might be mediated by the intrinsic resistance of senescent cells to apoptosis in combination with an impaired clearance of senescent cells by the immune system, which is required for limiting the retention of senescent cells in tissues.Citation2-5 The clearance, which is mediated by natural killer (NK) cells in a number of pathologies, is a tightly regulated process with variable efficacy.Citation4,5 Therefore, the intrinsic mechanisms that maintain the viability of senescent cells play a central role in the regulation of the presence of senescent cells in the organism.
Surprisingly, increased levels of p21, which promote cell cycle arrest in senescent cells, also maintain their viability, especially when senescence is driven by DNA-damage.Citation6 Indeed, persistent DNA damage response (DDR) is a central molecular mechanism that mediates senescence in aging and age-related diseases and induces up-regulation of p21.Citation2 Importantly, p21 knockdown of senescence cells enhances DDR as a result of the attempt of these cells to re-enter the cell-cycle (). This enhancement is accompanied by an increase in the activation of ATM (ataxia telangiectasia mutated), and nuclear factor (NF)-κB kinases. NF-κB activation induces TNF-α secretion and JNK activation to mediate death of senescent cells in a caspase- and JNK-dependent manner.Citation6 Knockdown of p21 in senescent cells can activate several modes of cell death involving both caspases and JNK. For instance, JNK activation might shift the balance of TNF-stimulated cell death from apoptosis to necrosis. Indeed, senescent cells present a significant elevation in TNF-α production and JNK phosphorylation following p21 knockdown, but not die following JNK inhibition.Citation6 Further research is necessary in order to define the mode of cell death induced in senescent cells after knockdown of p21.
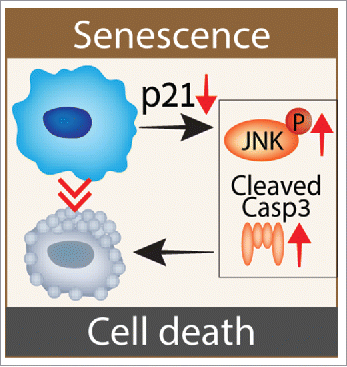
Figure 1. p21 maintains the viability of DNA damage-induced senescent (DIS) cells in a caspase-dependent and JNK-dependent manner. Following p21 knockdown, senescent cells attempt to enter the cell cycle, but fail and activate DNA damage response and NF-κB pathways.Citation6 This activation further leads to JNK-phosphorylation and cleavage of the pro-apoptotic Caspase-3, which are necessary for cell death induction by p21 knockdown.
The increase in p21 level also maintains the presence of senescent cells in pathological conditions in vivo. For example, in liver fibrosis activated hepatic stellate cells (HSC) activate DDR and become senescent, in order to restrain liver fibrosis and return the liver to a pre-damaged state.Citation2,5 The presence of senescent cells in a fibrotic liver is diminished in p21−/− mice.Citation6 This reduction might result from increased death of senescent cells, or alternatively, their decreased formation owing to induction of apoptosis following DDR activation in these cells. Of note, p21−/− mice develop less fibrosis then wild type mice as a result of a combination of cell autonomous and cell non-autonomous effects of p21 knockout. In a cell-autonomous manner, the inhibitory effect of p21 knockout on Transforming Growth Factor (TGF)-β signaling and collagen production in HSCs directly leads to reduced collagen deposition and fibrosis. Alternatively, p21 knockout increases TNF-α production in senescent cells. TNF-α can affect the activation of HSCs in a cell non-autonomous manner via its ability to antagonize the pro-fibrotic effect of TGF-β. It is also possible that the effect of p21 knockout on liver fibrosis stems from its ability to reduce the viability of senescent cells and to regulate the microenvironment by directly controlling the production of ECM components. Notably, TGF-β and TNF-α interplay regulates fibrotic processes in many tissues in the organism. Therefore, p21 inhibition not only leads to the elimination of senescent cells, it might also limit fibrotic processes in different tissues and promote tissue regeneration.
In addition to limiting tissue damage, senescent cell elimination can increase the proliferative capacity of stem and progenitor cells and hence support tissue regeneration.Citation3 Interestingly, p21 knockout was previously shown to support tissue regeneration and to limit aging phenotypes through various mechanisms.Citation7 Therefore, we suggest that rapid elimination of senescent cells following p21 silencing may not only limit the damage, induced by the presence of senescent cells, but also promote a better recovery of tissues due to an increase in its regenerative capacity. Inhibition of p21 may provide an efficient way to treat both fibrotic and non-fibrotic age-related pathologies. Overall, p21 silencing, reveals central molecular mechanisms responsible for maintaining the viability and retention of senescent cells in tissues, and suggests that the elimination of senescent cells by inhibition of these mechanisms represents a promising strategy to target senescent cells in order to promote tissue fitness and extend health-span and lifespan.
References
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400-14. doi:10.1038/nrc2657. PMID:19440234
- Burton, DG, Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. 2014;71(22):4373-4386. doi:10.1007/s00018-014-1691-3. PMID:25080110
- Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Communications. 2016;7:11190. doi:10.1038/ncomms11190.
- Biran A, Perelmutter M, Gal H, Burton DG, Ovadya Y, Vadai E, Geiger T, Krizhanovsky V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015;29:791-802. doi:10.1101/gad.259341.115. PMID:25854920
- Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging. 2016;8:328-344. doi:10.18632/aging.100897. PMID:26878797
- Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, Krizhanovsky V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. Embo J. 2017;36(15):2280-2295; e201695553. doi:10.15252/embj.201695553. PMID:28607003
- Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genetics. 2006;39:99-105. doi:10.1038/ng1937. PMID:17143283