
ABSTRACT
Autophagy is critical for homeostasis and cell survival during stress, but can also lead to cell death, a little understood process that has been shown to contribute to developmental cell death in lower model organisms, and to human cancer cell death. We recently reportedCitation1 on our thorough molecular and morphologic characterization of an autophagic cell death system involving resveratrol treatment of lung carcinoma cells. To gain mechanistic insight into this death program, we performed a signalome-wide RNAi screen for genes whose functions are necessary for resveratrol-induced death. The screen identified GBA1, the gene encoding the lysosomal enzyme glucocerebrosidase, as an important mediator of autophagic cell death. Here we further show the physiological relevance of GBA1 to developmental cell death in midgut regression during Drosophila metamorphosis. We observed a delay in midgut cell death in two independent Gba1a RNAi lines, indicating the critical importance of Gba1a for midgut development. Interestingly, loss-of-function GBA1 mutations lead to Gaucher Disease and are a significant risk factor for Parkinson Disease, which have been associated with defective autophagy. Thus GBA1 is a conserved element critical for maintaining proper levels of autophagy, with high levels leading to autophagic cell death.
Introduction
Autophagy is a catabolic process in which double-membrane vesicles, called autophagosomes, are formed de novo to engulf cytoplasm and organelles, eventually fusing with the lysosome to form the autolysosome, within which the internal contents are degraded.Citation2,3 Induction of autophagy is tightly regulated by the mTOR and Ulk1/2 kinases and their various modulators, which control the activation of the Vps34 phosphatidylinositol 3-kinase (PI3K). The PI3K complex, which includes Vps34, Beclin 1 and Atg14, produces PI3P at the sites of membrane nucleation, thereby recruiting the remaining Atg proteins. These proteins drive sequential ubiquitin-like conjugation reactions that ultimately produce membrane bound, lipidated MAP1B-LC3 (or LC3, orthologue of yeast Atg8). All of these steps are regulated at multiple levels, including post-transcriptional modifications and transcriptional mechanisms.Citation4 There are also negative feedback loops that regulate autophagosome turnover, so that autophagy flux levels are finely tuned to match the cellular need. In the normal cell, autophagy occurs at low levels in a constitutive manner to enable degradation and recycling of long-lived proteins, and removal of damaged organelles and misfolded proteins that can form in the course of normal cell functioning.Citation5 As such, it is essential for maintaining homeostasis and preventing disease.
During cell stress, nutrient deprivation or growth factor withdrawal, autophagy is induced above basal levels by inactivation of mTOR and subsequent activation of Ulk1 and its targets within the PI3K Vps34/Beclin 1 complex.Citation3,6 The resulting enhanced autophagy flux is critical for the cell to mitigate the damage and overcome the stress until more tenable conditions are restored. This can be achieved, for example, by autophagy-mediated removal of accumulated unfolded proteins during ER stress, or by the recycling of macromolecules as a source of nutrients and molecular building blocks during starvation (for example, see ref. Citation7). In addition, autophagy eliminates damaged mitochondria by mitophagy, thereby 1. limiting radical oxygen species (ROS) formation and consequently, DNA damage, and also 2. directly preventing mitochondrial outer membrane permeabilization (MOMP) and the release of apoptogenic factors.Citation8-10 In these scenarios, autophagy ensures cell survival, and often blocks apoptotic cell death. When homeostasis is restored, autolysosomes are recycled, lysosomes are reformed, and autophagy flux readjusts to basal levels, by reactivation of mTOR and reduction in several Atg protein levels (e.g. Refs. Citation11,12,13). If these shut-down mechanisms fail to initiate, or if the induction steps are overly activated, autophagy flux levels can potentially reach a point where they are no longer beneficial, but instead indiscriminately consume cellular organelles and proteins until the cell can no longer function properly. Under such circumstances, autophagy would directly lead to cell death.
Autophagic cell death
While the importance of maintaining the appropriate levels of autophagy for each cell's circumstance is obvious, it is less clear whether autophagic cell death is a physiologically relevant process. In fact, for a long time the existence of autophagic cell death was considered controversial.Citation14,15 Firstly, many studies, especially earlier ones, claimed to have observed autophagic cell death when they were in fact observing cell death that was accompanied by, but not caused by, autophagy.Citation16 In these cases, autophagy was likely induced to counter the death stimulus and block apoptosis, thereby representing a failed attempt to rescue the cell. The premature and often erroneous conclusions of these correlative studies paradoxically led many researchers to question the very premise of autophagic cell death.
As the field progressed, knock-out (KO) or knock-down (KD)-based targeted disruption of autophagy-specific genes was achieved, so that the nature of cell death could be determined not only through morphological observations, but also via analysis of the effects of blocking autophagy on cell death. However, a second complicating factor emerged, as studies showed scenarios in which autophagy was necessary for cell death, but that it occurred in conjunction with other cell death pathways, such as apoptosis or necroptosis. Moreover, autophagic processes have been shown to facilitate apoptosis or necroptosis, for example, by providing a scaffold for caspase activation or necrosome assembly,Citation17-23 or by targeting anti-apoptotic or survival proteins for selective degradation.Citation22,24-26 In these cases, autophagy was necessary for cell death but was only an indirect cause, leading researchers to doubt that autophagy can kill directly.
In light of the above studies and the questions they raised, in order to substantiate authentic autophagic cell death, researchers must not only conclusively prove that the stimulus triggers an increase in autophagic flux in the absence of alternative death pathways, but also prove that inhibition of various autophagic proteins that drive different steps in autophagy protects from cell death.Citation15,27 Scenarios wherein these criteria are best met are during developmental cell death in simpler model organisms such as the slime mold Dictyostelium discoideum and fruitfly Drosophila melanogaster. Under starvation conditions, and in response to cAMP, unicellular Dictyostelium aggregate into a multi-cellular organism that undergoes autophagy-dependent morphogenesis to form a spore-producing fruiting body. While not directly causing cell death, autophagy primes the cells so that when they are exposed to a second signal- the stalk Differentiation-Inducing Factor DIF-1- autophagic cell death results.Citation28,29 Similarly, removal of the larval midgut and salivary glands during Drosophila metamorphosis occurs by an apoptosis-independent cell death that requires autophagy genes, with specific components of the autophagy pathway differentially required for each process.Citation30-32 (see also ref. Citation33 for review) However, complete removal of the salivary glands requires the concerted actions of both autophagy and apoptosis.Citation30
Studies in higher mammals are more limited, since it is technically unfeasible to assess the effects of KO of multiple ATG genes on the death phenotype, due to early lethality and/or pathologies that develop even in tissue-specific ATG KOs, such as in the brain.Citation34 Yet, through the use of limited genetic manipulations and drugs that affect the pathway, autophagic cell death has been implicated in adult and neonatal animal models of cerebral hypoxia/ischemic injury,Citation35-39 and reperfusion injury of the heart.Citation40 Autophagic cell death has also been shown in cell culture models, which are more amenable to genetic manipulations. Cancer cells seem to be particularly susceptible to apoptosis-independent autophagic cell death, which was observed in response to hypoxia and oxidative stress, and to various anti-cancer drugs such as resveratrol, BH3 mimetics and betulinic acid (reviewed in ref. Citation41). Likewise, excessive autophagic consumption of damaged mitochondria, or mitophagy, led to melanoma cell death upon activation of the orphan nuclear receptor TR3.Citation42
Thus, while autophagic cell death has been clearly demonstrated, the field has still not advanced due to a lack of detailed morphological analysis and limited molecular characterization of the process. We still do not understand what switches the tightly regulated autophagy pathway from functioning as a survival pathway, to becoming a lethal one. In an initial attempt to address this issue, an inhibitor screen was performed to identify modulators of a specific form of autophagic cell death, termed autosis, which was induced by a cell-permeable peptide activating Beclin 1.Citation43 Significantly, this death pathway was confirmed in pathophysiological settings, such as starvation in cell culture, hypoxia-ischemia in rat brain, and aneroxia-nervosa in human patients.Citation43,44 The study showed that autosis was independent of apoptosis or necroptosis, and while it required ATG genes and autophagosome formation, autophagy flux was actually stalled at the degradative stage. Autosis was blocked by an inhibitor of the Na+/K+-ATPase ion channel, suggesting that cell death was a secondary response to stalled autophagy flux by a mechanism involving changes in ion transport and/or cellular osmolarity.Citation43 This study was an encouraging step in the direction of understanding an autophagy-dependent cell death, albeit an alternative form that did not involve autophagy flux.
Defining a model system of autophagic cell death induced by resveratrol
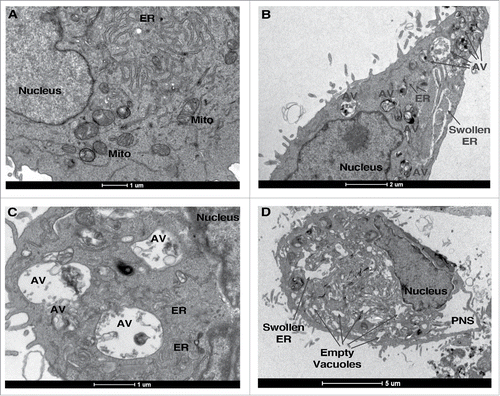
We recently attempted to define a system of true autophagic cell death that fulfilled the strict criteria stated above in order to characterize the phenomenon at the morphological level, and to identify molecular regulators of the process.Citation1 After detailed analysis of the death phenotype, we chose resveratrol (RSV) treatment of A549 lung carcinoma cells as the model system for further study. In these cells, RSV induced a time- and concentration-dependent cell death. None of the characteristic features of apoptosis or necroptosis were observed, and neither apoptosis nor necroptosis inhibitors rescued cell viability. In contrast, RSV induced abundant autophagosome accumulation with time, which we showed to represent active autophagy flux. Autophagic flux continued to increase for as long as 48 h, when cell death was most prominent. This feature clearly distinguishes the RSV-induced process from survival autophagy, such as that induced by starvation, which is transient: autophagy decreases to basal rates as the cell recovers. During RSV treatment, in contrast, the number of autophagosomes and autolysosomes continued to increase in time, to the point where they comprised most of the cytoplasmic area, and the cell was conspicuously devoid of organelles, with pronounced alterations in intracellular membranes (e.g. ). Thus autophagy was hyperactivated in this system. Most significantly, knock-down of several different ATG genes attenuated both autophagy and cell death, indicating that RSV-induced death requires autophagy, i.e., it is a bona fide form of autophagic cell death.
Figure 1. Resveratrol induces excessive autophagy in A549 cells. A. Transmission EM of control A549 cells. B-D. Transmission EM of A549 cells treated with 200 µM RSV for 48 h. AV, autophagic vacuoles, mito, mitochondria, PNS, perinuclear space.
Identifying GBA1 as a positive mediator of autophagic cell death
Using this system, we then conducted a signalome-wide shRNA screen for genes whose KD conferred a survival advantage to RSV-induced autophagic cell death.Citation1 The screen utilized Cellecta's Lentiviral shRNA Library Module 1, which targets more than 5000 signaling-related genes with multiple shRNAs/gene. RNA-seq was used to identify shRNAs that were enriched in surviving clones after prolonged exposure to RSV. Positive hits were genes that showed a statistically significant enrichment of greater than 50% for at least 3 independent shRNAs. These were further prioritized based on a sum of ranked standardization scores algorithm. The resulting set of genes was then validated by a siRNA-based secondary viability screen, finally yielding 8 candidate genes whose KD enhanced cell viability during RSV treatment by more than 50%.
One of the top hits on this short list was GBA1, the gene mutated in Gaucher Disease (GD). GBA1 encodes the lysosomal enzyme glucocerebrosidase, or acid β-glucosidase (GCase), which metabolizes glucosylceramide (GlcCer) to ceramide and glucose, comprising an important salvage pathway for ceramide generation. In fact, our data indicated that GCase protein and activity levels were increased upon RSV treatment in A549 cells at 48 h, concomitantly with enhanced autophagy.Citation1 This was accompanied by increased production of long-acyl chain ceramide species, and the sphingolipid derivatives sphingosine and sphingosine-1-phosphate. The increases in GCase levels correlated with the decreased viability that was observed in RSV-treated A549 cells. GBA1 KD attenuated GCase activity and ceramide production, and consistent with the screen results, rescued autophagic cell death. The number of cytoplasmic autophagic vesicles normally observed at late time-points of RSV treatment was reduced upon GCase KD, and at the ultrastructural level, GBA1 KD improved overall cell morphology, including that of the mitochondria, ER and Golgi. Interestingly, GCase was not elevated during starvation-induced autophagy in A549 cells, suggesting that it is not a general regulator of autophagy, but specifically plays a role in scenarios wherein autophagy leads to cell death.
Altogether, our data clearly indicated a role for GCase and ceramide production in autophagic cell death in our model system. Notably, the connection between ceramide and autopagic cell death was also suggested by a recent study in which treatment of a cell culture model of myeloid-derived suppressor cells with an inhibitor of acid ceramidase led to enhanced C16-ceramide production, autophagosome accumulation, and ultimately, apoptosis- and necroptosis-independent cell death.Citation45 Intriguingly, enhanced GBA1 expression levels and enzymatic activity have been reported in placenta from pregnancies complicated by preeclampsia, a disorder involving hypoxia, oxidative stress, and trophoblast cell death, which jeopardizes both mother and fetus.Citation46 Moreover, levels of sphingolipids and various ceramide species were observed to be elevated in preeclampsia placenta and proven to induce autophagy in trophoblasts.Citation47 This latter study attributed ceramide production to de novo synthesis and reduced lysosomal hydrolase activity, but did not consider the contribution of GBA1. While both apoptosis (e.g. ref. Citation48) and necroptosisCitation49 have been observed in preeclampsia placenta, there have also been reports of increased LC3-II and autophagosome formation in both placenta from preeclampsia patients, and in cultured hypoxic trophoblasts.Citation50 More comprehensive studies are needed to determine if this represents enhanced active autophagy flux, and whether its function is to promote cell survival or death. Considering our data, it would be very interesting to determine if autophagic cell death occurs during preeclampsia, and whether it is GBA1-dependent.
Proving the physiological relevance of GBA1 to developmental cell death in midgut regression of Drosophila larva
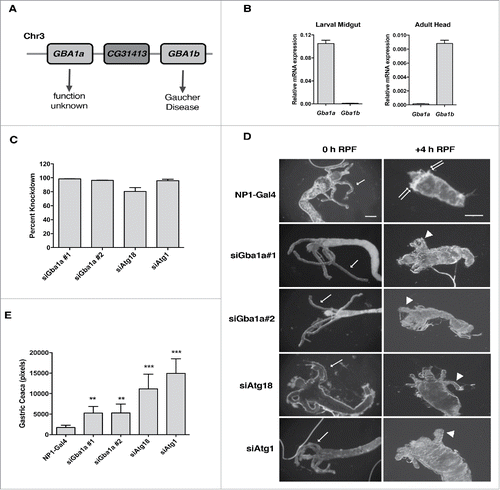
We wished to conclusively establish GBA1's function in a validated physiological scenario of autophagy and autophagic cell death. To this end, we examined the contribution of GBA1 to larval midgut cell death during Drosophila developmental metamorphosis, a critical period during which most of the larval tissues that were produced during embryogenesis are destroyed, and adult body structures are formed. While most of the cell death associated with Drosophila metamorphosis is apoptotic, destruction of the larval midgut requires autophagy and is caspase-independent.Citation31 The human GBA1 gene has two orthologues in Drosophila melanogaster, referred to as Gba1a (CG31148) and Gba1b (CG31414), respectively. The two Gba1 orthologues reside on chromosome 3, interposed by the CG31413 gene (). Expression data from the online resources modENCODECitation51 and FlyAtlasCitation52 showed that Gba1a is almost exclusively expressed in larval and adult midguts, whereas Gba1b is highly expressed throughout the developmental and adult stages in a wide variety of tissues and organs. Gba1b has been associated with fly models of GD.Citation53,54 We confirmed the expression data by quantitative real-time PCR analysis of the Gba1 orthologues: Gba1a was expressed in larval midgut at high levels in comparison to Gba1b, while conversely, Gba1b showed high expression in adult head, where Gba1a was barely detectable (). Thus Gba1a is likely to be the relevant orthologue for studying autophagic cell death of the larval midgut. RNA interference (RNAi) was used to knock-down expression of Gba1a, and as positive controls, the autophagy genes Atg1 and Atg18, specifically in the midgut, using the midgut driver NP1-Gal4 (Myo31DF-Gal4)Citation55 (see Materials and Methods for further detail). Nearly full KD efficiencies of the RNAi lines were demonstrated by quantitative real-time PCR analysis (). Larval midgut cell death is initiated at the end of the third instar larval stage and the beginning of white pupa formation, referred to as 0 h relative puparium formation (RPF). The midgut undergoes a drastic regression from 0 h RPF to +4 h RPF, which can be closely followed by observing the elongated gastric caeca present at the junction of the foregut and midgut, just below the bud-like proventriculus (, arrows in left-hand panels). The gastric caeca present at 0 h RPF begin to regress and are removed by +4 h RPF (, see top panel, right), whereas the remaining midgut tissues gradually regress over the next 12–24 h (not shown). As previously reported,Citation31 midgut-specific KD of the autophagy genes Atg1 and Atg18 resulted in a pronounced delay in midgut regression, evidenced by persistent gastric caeca at +4 h RPF, when compared to the control NP1-Gal4 line (, ). Importantly, morphological analysis of midguts at +4 h RPF in two independent Gba1a RNAi lines revealed a significant delay in midgut cell death, with only partial regression of the gastric caeca (, ). These data indicate that similar to autophagy gene KD, Gba1a KD had a significant inhibitory effect on midgut regression during the larval to early pupal transition, albeit not as strong as that of the central regulators of the process. This data imply a physiological role for Gba1a in developmental autophagic cell death.
Figure 2. Gba1a is necessary for midgut regression in Drosophila larva. (A) Schematic representation of the Gba1a and GBa1b gene loci on Drosophila chromosome 3. (B) Expression levels of Gba1a and Gba1b in larval midgut and adult head were determined by quantitative real-time PCR. Assays were performed in triplicate and normalized to RpL32/rp49 as an internal control. (C) Percent knockdown of Gba1a, Atg1 and Atg18 in the late third instar larvae (−4 h to −1 h RPF). The indicated siRNA constructs were expressed in larvae via the midgut driver NP1-Gal4. Transcript levels were measured by quantitative RT-PCR, normalized to the internal control RpL32/rp49, and are presented as relative KD of expression in comparison to expression in the NP1-Gal4 line. (D) Morphology of midguts at 0 h RPF and +4 h RPF comparing control (NP1-Gal4) pupae with pupae from NP1-Gal4>UAS-Gba1a RNAi #1 (siGba1a#1), NP1-Gal4>UAS-Gba1a RNAi #2 (siGba1a#2), NP1-Gal4>UAS-Atg1 RNAi (siAtg1) and NP1-Gal4>UAS-Atg18 RNAi (siAtg18) crosses. Arrows indicate gastric caeca at 0 h; double arrows, normal regression of gastric caeca at 4 h; arrowheads, persistent gastric caeca in KD larvae. Scale bar represents 200 µm. (E) Quantitation of gastric caeca size at +4 h RPF. Data represents mean ± SD of more than 10 midguts per genotype; statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Tukey's post hoc test, **P < 0.01; ***P < 0.001.
GBA1 dysfunction in human disease
In humans, homozygous loss-of-function GBA1 mutations lead to GD, a lysosomal storage disease in which GlcCer accumulates as a result of the deficiency in GCase and the subsequent inability of the lysosome to hydrolyze GlcCer to ceramide.Citation56 GD is currently treated by a combination of enzyme replacement therapy with recombinant GCase and substrate reduction therapy. More recently, GBA1 mutations have also been found to be an increased risk factor for Parkinson Disease (PD), and have been associated with accumulation of α-synuclein aggregates as a result of defective autophagy-lysosomal function; the level of functional GCase produced from the mutated gene correlates with severity of disease/disease risk.Citation57 Significantly, KO of GBA1 (i.e. Gba1b) in Drosophila brains led to GlcCer accumulation and lysosomal pathology similar to GD, and neurodegeneration and synaptic loss, similar to PD.Citation53,54 Furthermore, an increase in autophagosome number and a block in autophagy flux were observed, consistent with defects at the autolysosome stage. These observations demonstrate a strong conservation of Gba1 functional roles in the fly. Autophagy defects and accumulation of α-synuclein inclusions were also observed in other models of inhibition of GCase by drugs, and KD or KO of GBA1 in mice, MEFs and cell culturesCitation58-61 (also reviewed in ref. Citation62). Thus, GCase is critical for functional autophagy and prevention of neurodegeneration.
Conclusion and perspective
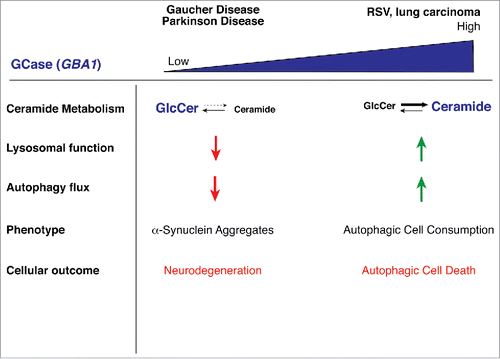
The studies described herein demonstrate the critical role that GBA1 plays in the regulation of autophagy, and that both loss-of-function and gain-of-function are detrimental to the balance of autophagy within the cell (). Too little GCase activity leads to the inability of the cellular autophagy pathway to deal with aggregating α-synuclein, with dire consequences that lead to neuropathy. Too much GCase converts a beneficial pathway into a lethal one, whereby the cell literally consumes itself to death. The question still remains how changes in sphingolipid metabolism and/or increased ceramide production directly cause autophagic cell death. It may have to do with differences in membrane properties and curvature that support enhanced autophagosome formation and maturation, or lysosomal recycling. Future studies that address these issues will not only further enhance our understanding of autophagic cell death, but may also be applicable to GBA1-related pathologies such as GD and PD.
Figure 3. Model showing effects of different expression levels of GBA1. Loss of GBA1 expression, such as that which results from mutations associated with Gaucher Disease or Parkinson Disease, leads to GlcCer accumulation, lysosomal dysfunction, defects in autophagy, and eventual accumulation of α-synuclein inclusions, resulting in neurodegeneration. Enhanced GBA1 expression, such as that observed following RSV treatment, leads to elevations in ceramide and its sphingolipid metabolites, induction of autophagy and autophagic cell death.
Materials and methods
Drosophila genetics
All fly lines were maintained on cornmeal, molasses and yeast medium at 25°C. RNAi-based knockdown of specific genes was achieved by crossing midgut driver (NP1-Gal4) flies (Drosophila Genetic Resource Center, Kyoto (DGRC #112001)) to flies bearing UAS-RNAi lines (obtained either from the Vienna Drosophila Resource Center (VDRC) or the Bloomington Drosophila Stock Center (BDSC) at Indiana University). UAS-RNAi lines used in the study include Gba1a#1 (BDSC #39064), Gba1a#2 (VDRC #GD14697), Atg18 (BDSC #28061) and Atg1 (BDSC #26731). All flies were maintained and crossed at 25°C. Larva were fed with 0.05% bromophenol blue containing food and staged by observing the midgut retention or clearing of bromophenol blue dye. Wandering clear gut larva were transferred onto a damp Whatmann paper placed in a petri dish and dissected as previously described.Citation63
Gene expression analysis
RNA was extracted from 30 whole heads from 5 day old adult flies and 20–30 midguts from late third instar larvae of the indicated genotypes, using TRIzol (Life Technologies). cDNA was generated using High Capacity RNA-to-cDNA Kit (Applied Biosystems). Quantitative real-time PCR based quantitation of Gba1a, Gba1b, Atg1, Atg18a and RpL32/rp49 mRNA expression was performed using primers published elsewhere.Citation32,54 Quantitative real-time PCR was performed using FAST SYBR Green Mater Mix (Applied Biosystems). Assays were performed in triplicates, normalized to RpL32/rp49, and analyzed according to the comparative Ct method.Citation64
Midgut morphology and quantitation
Animals were staged and collected at −4 h RPF (crawling 3rd instar larvae with everted spiracles), 0 h RPF (white prepupae) or +4 h RPF (white prepupae aged for 4 h). Midguts were dissected in PBS, fixed in 4% paraformaldehyde in PBS, mounted onto glass slides using IMMU-MOUNT (Thermo Scientific) and imaged using a stereozoom microscope (Olympus). Digital images were obtained using a DP50 CCD camera with StudioLite software (Olympus). Measurements of gastric caeca size were performed using ImageJ software for more than 10 midguts per genotype. Gastric caeca edges were traced and pixel densities were determined using the histogram tool.
Statistical analysis
The statistical significance of differences between means was assessed by one-way ANOVA followed by Tukey's post hoc test. Values with P < 0.05 were considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the labs of Prof. Talila Volk and Prof. Benny Shilo for allowing us to use their Drosophila lab facilities and Unnikannan Cp for his technical help.
Funding
This work was supported by a grant from the European Research Council under the European Union's Seventh Framework Program (FP7/2007-2013/ERC, grant agreement 322709).
References
- Dasari SK, Bialik S, Levin-Zaidman S, Levin-Salomon V, Merrill AH, Jr., Futerman AH, Kimchi A. Signalome-wide rnai screen identifies gba1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017;24(7):1288-1302. https://doi.org/10.1038/cdd.2017.80. PMID:28574511
- Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124-31. https://doi.org/10.1016/j.ceb.2009.11.014. PMID:20034776
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Mammalian autophagy: How does it work? Annu Rev Biochem. 2016;85:685-713.
- Botti-Millet J, Nascimbeni AC, Dupont N, Morel E, Codogno P. Fine-tuning autophagy: From transcriptional to posttranslational regulation. Am J Physiol Cell Physiol. 2016;311(3):C351-362. https://doi.org/10.1152/ajpcell.00129.2016. PMID:27335173
- Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147(4):728-41. https://doi.org/10.1016/j.cell.2011.10.026. PMID:22078875
- Meijer AJ, Lorin S, Blommaart EF, Codogno P. Regulation of autophagy by amino acids and mtor-dependent signal transduction. Amino Acids. 2015;47(10):2037-63. https://doi.org/10.1007/s00726-014-1765-4. PMID:24880909
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237-48. https://doi.org/10.1016/j.cell.2004.11.046. PMID:15680329
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795-800. https://doi.org/10.1101/gad.2016211. PMID:21498569
- Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila). 2011;4(7):973-83. https://doi.org/10.1158/1940-6207.CAPR-10-0387. PMID:21733821
- Bhogal R, Weston C, Curbishley S, Adams D, Afford S. A cyto-protective mechanism which prevents primary human hepatocyte apoptosis during oxidative stress. Autophagy. 2012;8(4):1-14. https://doi.org/10.4161/auto.19012. PMID:22082964
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of ampk-mtor-ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2-11. https://doi.org/10.1128/MCB.06159-11. PMID:22025673
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, et al. Cul3-klhl20 ubiquitin ligase governs the turnover of ulk1 and vps34 complexes to control autophagy termination. Mol Cell. 2016;61(1):84-97. https://doi.org/10.1016/j.molcel.2015.11.001. PMID:26687681
- Feng Y, Klionsky DJ. Downregulation of autophagy through cul3-klhl20-mediated turnover of the ulk1 and pik3c3/vps34 complexes. Autophagy. 2016;12(7):1071-72. https://doi.org/10.1080/15548627.2016.1173802. PMID:27096860
- Shen H-M, Codogno P. Autophagic cell death: Loch ness monster or endangered species? Autophagy. 2011;7(5):457-65. https://doi.org/10.4161/auto.7.5.14226. PMID:21150268
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012;19(1):87-95. https://doi.org/10.1038/cdd.2011.146. PMID:22052193
- Kroemer G, Levine B. Autophagic cell death: The story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004-10. https://doi.org/10.1038/nrm2529. PMID:18971948
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (idisc)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287(15):12455-468. https://doi.org/10.1074/jbc.M111.309104. PMID:22362782
- Laussmann MA, Passante E, Dussmann H, Rauen JA, Wurstle ML, Delgado ME, Devocelle M, Prehn JHM, Rehm M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011;18(10):1584-97. https://doi.org/10.1038/cdd.2011.27. PMID:21455219
- Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al. Essential roles of atg5 and fadd in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280(21):20722-729. https://doi.org/10.1074/jbc.M413934200. PMID:15778222
- Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721-35. https://doi.org/10.1016/j.cell.2009.03.015. PMID:19427028
- Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37(4):337-49. https://doi.org/10.1016/j.devcel.2016.04.018. PMID:27219062
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103(13):4952-57. https://doi.org/10.1073/pnas.0511288103. PMID:16547133
- Basit F, Cristofanon S, Fulda S. Obatoclax (gx15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20(9):1161-73. https://doi.org/10.1038/cdd.2013.45. PMID:23744296
- Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dbruce controls DNA fragmentation in nurse cells during late drosophila melanogaster oogenesis. J Cell Biol. 2010;190(4):523-31. https://doi.org/10.1083/jcb.201002035. PMID:20713604
- Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of fap-1. Nat Cell Biol. 2014;16(1):47-54. https://doi.org/10.1038/ncb2886. PMID:24316673
- He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, et al. A jnk-mediated autophagy pathway that triggers c-iap degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33(23):3004-13. https://doi.org/10.1038/onc.2013.256. PMID:23831571
- Shen HM, Codogno P. Autophagic cell death: Loch ness monster or endangered species? Autophagy. 2011;7(5):457-65. https://doi.org/10.4161/auto.7.5.14226. PMID:21150268
- Giusti C, Luciani MF, Golstein P. A second signal for autophagic cell death? Autophagy. 2010;6(6):823-24. https://doi.org/10.4161/auto.6.6.12750. PMID:20639697
- Calvo-Garrido J, Carilla-Latorre S, Kubohara Y, Santos-Rodrigo N, Mesquita A, Soldati T, Golstein P, Escalante R. Autophagy in dictyostelium: Genes and pathways, cell death and infection. Autophagy. 2010;6(6):686-701. https://doi.org/10.4161/auto.6.6.12513. PMID:20603609
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in drosophila. Cell. 2007;131(6):1137-48. https://doi.org/10.1016/j.cell.2007.10.048. PMID:18083103
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in drosophila. Curr Biol. 2009;19(20):1741-46. https://doi.org/10.1016/j.cub.2009.08.042. PMID:19818615
- Xu T, Nicolson S, Denton D, Kumar S. Distinct requirements of autophagy-related genes in programmed cell death. Cell Death Differ. 2015;22(11):1792-1802. https://doi.org/10.1038/cdd.2015.28. PMID:25882046
- Tracy K, Baehrecke EH. The role of autophagy in drosophila metamorphosis. Curr Top Dev Biol. 2013;103:101-125.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823-30. https://doi.org/10.1038/ncb0910-823. PMID:20811354
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454-69. https://doi.org/10.2353/ajpath.2008.070876. PMID:18187572
- Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG. Rna interference-mediated downregulation of beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin. 2009;30(7):919-27. https://doi.org/10.1038/aps.2009.79. PMID:19574998
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66(3):378-89. https://doi.org/10.1002/ana.21714. PMID:19551849
- Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, Li H, Li Q, Wang X, Uchiyama Y, et al. Neuroprotection by selective neuronal deletion of atg7 in neonatal brain injury. Autophagy. 2016;12(2):410-23. https://doi.org/10.1080/15548627.2015.1132134. PMID:26727396
- Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014;10(5):846-60. https://doi.org/10.4161/auto.28264. PMID:24674959
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of amp-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914-22. https://doi.org/10.1161/01.RES.0000261924.76669.36. PMID:17332429
- ulda S, Kogel D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34(40):5105-13. https://doi.org/10.1038/onc.2014.458. PMID:25619832
- Wang WJ, Wang Y, Chen HZ, Xing YZ, Li FW, Zhang Q, Zhou B, Zhang HK, Zhang J, Bian XL, et al. Orphan nuclear receptor tr3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol. 2014;10(2):133-40. https://doi.org/10.1038/nchembio.1406. PMID:24316735
- Liu Y, Shoji-Kawata S, Sumpter RM, Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. Autosis is a na+,k+-atpase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364-371. https://doi.org/10.1073/pnas.1319661110. PMID:24277826
- Kheloufi M, Boulanger CM, Codogno P, Rautou PE. Autosis occurs in the liver of patients with severe anorexia nervosa. Hepatology. 2015;62(2):657-8. https://doi.org/10.1002/hep.27597. PMID:25376517
- Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, Marshall B, Schoenlein PV, Lebedyeva IO, Liu K. Ceramide activates lysosomal cathepsin b and cathepsin d to attenuate autophagy and induces er stress to suppress myeloid-derived suppressor cells. Oncotarget. 2016;7(51):83907-25. PMID:27880732
- Jebbink JM, Boot RG, Keijser R, Moerland PD, Aten J, Veenboer GJ, van Wely M, Buimer M, Ver Loren van Themaat E, Aerts JM, et al. Increased glucocerebrosidase expression and activity in preeclamptic placenta. Placenta. 2015;36(2):160-9. https://doi.org/10.1016/j.placenta.2014.12.001. PMID:25552189
- Melland-Smith M, Ermini L, Chauvin S, Craig-Barnes H, Tagliaferro A, Todros T, Post M, Caniggia I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy. 2015;11(4):653-69. https://doi.org/10.1080/15548627.2015.1034414. PMID:25853898
- Leung DN, Smith SC, To KF, Sahota DS, Baker PN. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol. 2001;184(6):1249-50. https://doi.org/10.1067/mob.2001.112906. PMID:11349196
- Bailey LJ, Alahari S, Tagliaferro A, Post M, Caniggia I. Augmented trophoblast cell death in preeclampsia can proceed via ceramide-mediated necroptosis. Cell Death Dis. 2017;8(2):e2590. https://doi.org/10.1038/cddis.2016.483. PMID:28151467
- Gong JS, Kim GJ. The role of autophagy in the placenta as a regulator of cell death. Clin Exp Reprod Med. 2014;41(3):97-107. https://doi.org/10.5653/cerm.2014.41.3.97. PMID:25309853
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. The developmental transcriptome of drosophila melanogaster. Nature. 2011;471(7339):473-9. https://doi.org/10.1038/nature09715. PMID:21179090
- Chintapalli VR, Wang J, Dow JA. Using flyatlas to identify better drosophila melanogaster models of human disease. Nat Genet. 2007;39(6):715-20. https://doi.org/10.1038/ng2049. PMID:17534367
- Kinghorn KJ, Gronke S, Castillo-Quan JI, Woodling NS, Li L, Sirka E, Gegg M, Mills K, Hardy J, Bjedov I, et al. A drosophila model of neuronopathic gaucher disease demonstrates lysosomal-autophagic defects and altered mtor signalling and is functionally rescued by rapamycin. J Neurosci. 2016;36(46):11654-70. https://doi.org/10.1523/JNEUROSCI.4527-15.2016. PMID:27852774
- Davis MY, Trinh K, Thomas RE, Yu S, Germanos AA, Whitley BN, Sardi SP, Montine TJ, Pallanck LJ. Glucocerebrosidase deficiency in drosophila results in alpha-synuclein-independent protein aggregation and neurodegeneration. PLoS Genet. 2016;12(3):e1005944. https://doi.org/10.1371/journal.pgen.1005944. PMID:27019408
- Hayashi S, Ito K, Sado Y, Taniguchi M, Akimoto A, Takeuchi H, Aigaki T, Matsuzaki F, Nakagoshi H, Tanimura T, et al. Getdb, a database compiling expression patterns and molecular locations of a collection of gal4 enhancer traps. Genesis. 2002;34(1–2):58-61. https://doi.org/10.1002/gene.10137. PMID:12324948
- Dandana A, Ben Khelifa S, Chahed H, Miled A, Ferchichi S. Gaucher disease: Clinical, biological and therapeutic aspects. Pathobiology. 2016;83(1):13-23. https://doi.org/10.1159/000440865. PMID:26588331
- Schapira AH. Glucocerebrosidase and parkinson disease: Recent advances. Mol Cell Neurosci. 2015;66(Pt A):37-42. https://doi.org/10.1016/j.mcn.2015.03.013. PMID:25802027
- Bae EJ, Yang NY, Lee C, Lee HJ, Kim S, Sardi SP, Lee SJ. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp Mol Med. 2015;47:e153.
- Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun. 2014;5:4755.
- Du TT, Wang L, Duan CL, Lu LL, Zhang JL, Gao G, Qiu XB, Wang XM, Yang H. Gba deficiency promotes snca/alpha-synuclein accumulation through autophagic inhibition by inactivated ppp2a. Autophagy. 2015;11(10):1803-20. https://doi.org/10.1080/15548627.2015.1086055. PMID:26378614
- Farfel-Becker T, Vitner EB, Kelly SL, Bame JR, Duan J, Shinder V, Merrill AH, Jr., Dobrenis K, Futerman AH. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic gaucher disease leads to neurodegeneration. Hum Mol Genet. 2014;23(4):843-54. https://doi.org/10.1093/hmg/ddt468. PMID:24064337
- Kinghorn KJ, Asghari AM, Castillo-Quan JI. The emerging role of autophagic-lysosomal dysfunction in gaucher disease and parkinson's disease. Neural Regen Res. 2017;12(3):380-4. https://doi.org/10.4103/1673-5374.202934. PMID:28469644
- Andres AJ, Thummel CS. Methods for quantitative analysis of transcription in larvae and prepupae. Methods Cell Biol. 1994;44:565-573.
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative c(t) method. Nat Protoc. 2008;3(6):1101-08. https://doi.org/10.1038/nprot.2008.73. PMID:18546601