
ABSTRACT
The ataxia-telangiectasia-mutated (ATM) and rad3-related (ATR) checkpoint pathway plays an essential role in modulating cellular responses to replication stress and DNA damage to maintain genomic stability. In various tumors, cancer cells have increased dependence on ATR signaling for survival, making ATR a promising target for cancer therapy. ATR inhibitors sensitize multiple tumor cell types to radiation and DNA-damaging agents, but application of an ATR inhibitor alone shows limited efficacy. In the present study, we investigated the role of epithelial-to-mesenchymal transition (EMT) and the EMT transcription factor ZEB1 in regulating cell sensitivity to the ATR inhibitor VE-821. We found that VE-821 induced EMT with concomitant ZEB1 upregulation and promoted migration in cells in which the anti-proliferative effect of VE-821 was limited. Knocking down ZEB1 using siRNA partially reversed VE-821-induced EMT, and sensitized cells to VE-821 via effective attenuation of migration and AKT/ERK signaling. Moreover, ZEB1 inhibition promoted Chk1 phosphorylation and induced S-phase arrest by enhancing TopBP1 expression, which suggests a distinctive modulatory effect of ZEB1 on Chk1. Finally, combining VE-821 with ZEB1 inhibition enhanced DNA damage accumulation. These results demonstrate that EMT represents a novel mechanism for limiting the effectiveness of an ATR inhibitor, and thus suggest that ZEB1 inhibition might represent a new approach to increasing the efficiency of, or reversing resistance to, ATR inhibitors.
Introduction
Tumor cells require checkpoints to halt the cell cycle and promote DNA damage repair. Therefore, checkpoints are necessary to maintain genomic integrity during cancer cell proliferation [Citation1,2]. For this reason, checkpoint inhibition has emerged as a potential therapeutic target in cancer. In human cells, the ataxia-telangiectasia-mutated (ATM) kinase and the ATM and rad3-related (ATR) kinase are master regulators of two major checkpoint pathways [Citation3]. Whereas ATM primarily responds to double-stranded DNA breaks, ATR is activated by a wide spectrum of DNA damage and replication problems [Citation3,4]. Recently, more attention has focused on the ATR pathway because it is crucial for the repair of DNA replication stress and DNA damage during proliferation. Therefore, tumor cells have increased dependence on ATR signaling for survival, making ATR a promising target for cancer therapy [Citation1,2]
Although inhibiting ATR represents an attractive therapeutic strategy, the atypical nature of the phosphatidylinositol 3-kinase (PI3K)–related kinases (PIKKs), the family to which ATR belongs, has made developing a selective ATR inhibitor difficult. In 2011, the first potent and selective inhibitor, VE-821, was finally reported [Citation5]. Preclinical studies have shown that VE-821 and its analogue VE-822 (VX-970) robustly sensitize multiple tumor cell types to radiation and DNA-damaging agents including cisplatin, gemcitabine and topoisomerase I inhibitors in vitro [Citation5–9]. Similarly, in xenograft studies, ATR inhibitors have shown impressive results for sensitization of pancreatic cancer and colorectal cancer to radiation and chemotherapy [Citation7–9].
Treatment with a single ATR inhibitor exerted diverse anti-tumor activity in various cancers. However, VE-822 failed to reduce tumor growth in pancreatic tumor xenografts when used alone [Citation8]. Recently, another ATR inhibitor, AZD6738, was shown to exert anti-proliferative and DNA damage repair-inhibiting effects in HER2-positive breast cancer cells and ATM-deficient gastric cancer cells [Citation5,10]. Emerging evidence shows that checkpoint-defective tumors with loss of ATM and p53, DNA repair-defective tumors, or tumors with high replication stress may be particularly sensitive to ATR inhibition [Citation1,5,11]. Other mechanisms underlying tumor cell sensitivity to ATR inhibitors are less well understood and require investigation.
Among the diverse mechanisms of cancer therapeutic resistance, epithelial-to-mesenchymal transition (EMT) represents a major resistance mechanism to chemotherapy and radiotherapy in various cancers, and thus presents a major challenge in cancer research and treatment [Citation12,13]. However, whether EMT is associated with ATR inhibitors is unknown. As a driver of EMT, ZEB1 plays an important role in tumor progression and metastasis, and its expression correlates with poor clinical outcomes in cancer patients [Citation14,15]. Additionally, ZEB1 is involved in therapeutic resistance in multiple cancers. Previous studies suggested that ZEB1 mediated cisplatin resistance in ovarian cancer and gefitinib resistance in non-small-cell lung cancer [Citation16,17].
In this study, we report that upregulating ZEB1 during EMT improved the efficacy of the ATR inhibitor VE-821. Blocking ZEB1 partially reversed VE-821-induced EMT, which led to effective inhibition of migration and enhanced DNA damage accumulation, and sensitized cells to VE-821. These results demonstrate a novel resistance mechanism to an ATR inhibitor, and could be applied to further improving ATR inhibitor efficacy in clinical applications.
Results
The ATR inhibitor VE-821 induces EMT accompanied by ZEB1 upregulation
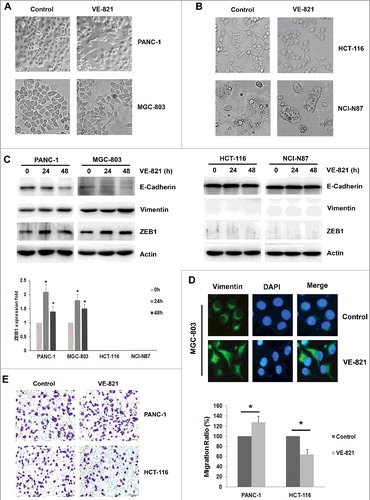
We chose four types of cancer cells that are dependent on the ATR/Chk1 pathway: three p53-deficient cancer cell lines (PANC-1, MGC-803 and NCI-N87) and a p53-wild-type/ATM-deficient cell line (HCT-116). When we applied the ATR inhibitor VE-821 to these four different cell lines, we observed distinct morphological changes. Unlike HCT-116 cells and NCI-N87 cells, some PANC-1 cells and MGC-803 cells changed from characteristic epithelial cells to spindle-shaped mesenchymal cells (, ). In keeping with this observation, downregulation of the epithelial marker E-cadherin and upregulation of vimentin were confirmed by western blotting and immunofluorescence staining in VE-821-treated PANC-1 and MGC-803 cells (, D). We then analyzed the common mesenchymal markers ZEB1, ZEB2, Twist2, Snail and Slug in these cells, and found that only the transcription factor ZEB1 exhibited increased expression levels concomitant with EMT in both cell types (, Supplementary Fig S1A, B). We next examined the effect of VE-821 on the cellular migration ability of PANC-1 and HCT-116 cells. Consistent with the observed morphological changes, the migratory potential of PANC-1 cells increased after VE-821 application (from 100 ± 0% to 128 ± 12%, p = 0.030), whereas the migratory ability of HCT-116 cells decreased after VE-821 application (from 100 ± 0% to 64 ± 10%, p = 0.013) (). These results demonstrate for the first time that the ATR inhibitor VE-821 induced EMT in PANC-1 and MGC-803 cells, and that ZEB1 was the key mediator of VE-821-induced EMT.
Figure 1. The effect of ATR inhibitor VE-821 on EMT and migration ability in four kinds of cancer cells. (A, B) Four kinds of cancer cells (PANC-1, MGC-803, HCT-116 and NCI-N87) were treated with 5 μM VE-821 for 48 h. Photos of cellular morphology were taken at × 200 magnification. (C) Four kinds of cancer cells were treated with 5 μM VE-821 for 24 h and 48 h. The expression of E-cadherin, Vimentin and ZEB1 was performed by Western Blotting. Actin was used as loading control. The fold of ZEB1 expression was quantified by Image J. *p<0.05 vs. control 0 h. (D) MGC-803 cells were stained with antibodies to Vimentin (green) and nuclei was stained with DAPI. Images were captured by fluorescence microscopy at × 40 magnification. (E) PANC-1 cells and HCT-116 cells were performed the migration assays as described in Materials and methods. Photos of migrated cells were taken at × 200 magnification. Data are means ± SD in three independent experiments.
ZEB1 inhibition abrogates VE-821-induced EMT
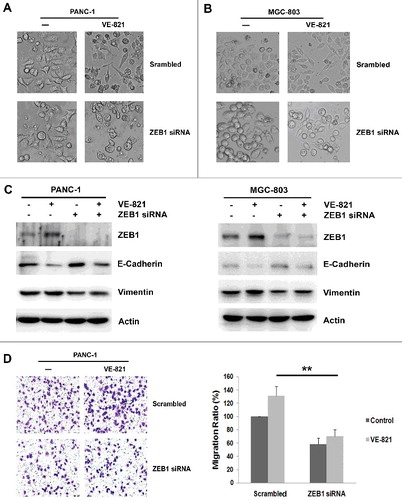
Next, we depleted ZEB1 expression using siRNA to explore the role of ZEB1 in VE-821-induced EMT. We observed a change in the morphology of cells treated with ZEB1 siRNA in combination with VE-821. As expected, ZEB1 inhibition apparently disrupted VE-821-induced changes to the epithelial–mesenchymal phenotype in both PANC-1 cells and MGC-803 cells (, ). Moreover, the downregulation of E-cadherin and upregulation of vimentin that occurred in cells exposed to VE-821 were partially reversed when ZEB1 expression was suppressed (). Furthermore, inhibition of ZEB1 expression repressed cell migratory ability, compared with treatment with a scrambled siRNA control (58.3 ± 9.1% vs. 100 ± 0%, p = 0.008) (). Similarly, ZEB1 inhibition further decreased the migratory potential of VE-821-treated PANC-1 cells (69.7 ± 10.8% vs. 131.1 ± 14.1%, p = 0.002) (). These results indicate that ZEB1 inhibition impaired VE-821-induced EMT, and reversed the VE-821-induced enhancement of migration.
Figure 2. ZEB1 inhibition reverses EMT induces by VE-821 and enhances migration ability. (A, B) PANC-1 cells and MGC-803 cells were transiently transfected with Scrambled Control siRNA or ZEB1 siRNA, then added with 5 μM VE-821 for 48 h. Photos of cellular morphology were taken at × 200 magnification. (C) PANC-1 cells and MGC-803 cells were transiently transfected with Scrambled Control siRNA or ZEB1 siRNA, then added with 5 μM VE-821 for 48 h. The expression of ZEB1, E-cadherin and Vimentin was performed by Western Blotting. (D) PANC-1 cells were transiently transfected with Scrambled Control siRNA or ZEB1 siRNA, then added with 5 μM VE-821 for 24 h. Then migration assays were performed and photos of migrated cells were taken at × 200 magnification. **p<0.01 vs. Scrambled Control.
ZEB1 inhibition attenuates AKT/ERK activation and sensitizes cells to the ATR inhibitor VE-821
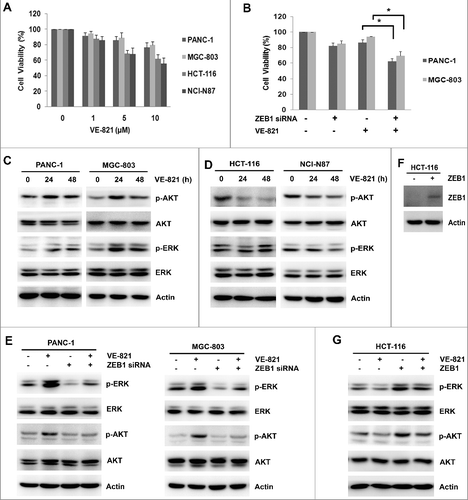
To investigate whether ZEB1 expression was involved in cell sensitivity to the ATR inhibitor VE-821, we tested the effects of ZEB1 knockdown on cell viability and cellular signaling in four VE-821-treated cell lines. The two cell lines that underwent EMT (PANC-1 and MGC-803) exhibited lower cell inhibition of VE-821 than the cells that did not undergo EMT (HCT-116 and NCI-N87) (). We next examined changes in proliferative signaling, because this also affects the EMT process. VE-821 decreased the basal level of phosphorylated AKT and phosphorylated ERK in HCT-116 cells and NCI-N87 cells, whereas it stimulated the expression of phosphorylated AKT and phosphorylated ERK in PANC-1 cells and MGC-803 cells induced to undergo EMT (, ). ZEB1 knockdown abolished the VE-821-induced activation of AKT and ERK in PANC-1 and MGC-803 cells (). Accordingly, cell viability was significantly reduced by combined ZEB1 knockdown and VE-821 treatment in PANC-1 cells (62.1 ± 4.0% vs. 86.3 ± 4.1%, p = 0.012) and MGC-803 cells (66.3 ± 5.7% vs. 88.6 ± 4.0%, p = 0.026) (). To further demonstrate the effect of ZEB1 on AKT and ERK, HCT-116 cells were transfected with pcDNA3.1/ZEB1-Flag plasmid to overexpress ZEB1 level (), and then added with VE-821. The results showed that ZEB1 overexpression abrogated inhibition of phosphorylated AKT and phosphorylated ERK by VE-821 in HCT-116 cells (). These results demonstrated that ZEB1 expression was the key factor regulating VE-821-induced EMT, and might contribute to the desensitization of cells to the anti- proliferative effect of VE-821.
Figure 3. ZEB1 inhibition increases sensitivity of VE-821 in PANC-1 cells and MGC-803 cells. (A) Indicated concentrations of VE-821 (0, 1, 5 and 10 μM) were added to four kinds of cancer cells (PANC-1, MGC-803, HCT-116 and NCI-N87) for 48 h. Cell viability was performed by MTT assay. Results from three independent experiments are shown. (B) PANC-1 cells and MGC-803 cells were transiently transfected with Scrambled Control siRNA or ZEB1 siRNA, then added with 5 μM VE-821 for 48 h. Cell viability was performed by MTT assay. *p<0.05 vs. VE-821 alone. (C, D) PANC-1, MGC-803, HCT-116 and NCI-N87 cells were added to 5μM VE-821 for 24 h and 48 h. The total and phosphorylation expression of AKT and ERK was performed by Western Blotting. (E) PANC-1 cells and MGC-803 cells were transiently transfected with Scrambled Control siRNA or ZEB1 siRNA, then added with 5 μM VE-821 for 24 h. The total and phosphorylation expression of AKT and ERK was performed by Western Blotting. (F) HCT-116 cells were transiently transfected with pcDNA3.1/ZEB1-Flag plasmid or pcDNA3.1 empty control, and expression of ZEB1 was performed by Western Blotting. (G) HCT-116 cells were transiently transfected with pcDNA3.1/ZEB1-Flag plasmid or pcDNA3.1 empty control, then added with 5 μM VE-821 for 24 h. The total and phosphorylation expression of AKT and ERK was performed by Western Blotting.
ZEB1 inhibition promotes Chk1 phosphorylation by enhancing TopBP1 expression, and induces S-phase arrest
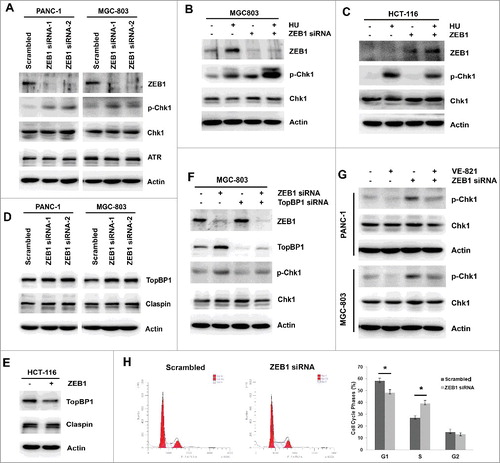
We next tested the effect of ZEB1 inhibition combined with VE-821 treatment on Chk1 phosphorylation and the cell cycle. First, the target of the ATR inhibitor VE-821 was examined by western blotting. The basal level of phosphorylated Chk1 was not apparently reduced by VE-821 for longer time (24 h and 48 h) in four cell lines (Supplemental Fig S2A, B). To assess the effect of ZEB1 inhibition on Chk1 phosphorylation and its mechanism, two pairs of siRNA were used to knockdown ZEB1 expression in both PANC-1 cells and MGC-803 cells. Following ZEB1 inhibition, Chk1 phosphorylation was elevated, whereas the total expression levels of Chk1 and ATR remained unchanged (). We then used HU to increase Chk1 phosphorylation as a positive control. As expected, combining HU treatment with ZEB1 inhibition further promoted Chk1 phosphorylation (). On the contrary, overexpression of ZEB1 attenuated HU-induced Chk1 phosphorylation (). To demonstrate the modulation of Chk1 phosphorylation by ZEB1, we examined TopBP1 and Claspin, two important adaptor proteins that facilitate Chk1 phosphorylation in response to genotoxic stress. When ZEB1 expression was suppressed, total TopBP1 expression was elevated in both PANC-1 cells and MGC-803 cells, while ZEB1 expression was elevated, TopBP1 expression was reduced in HCT-116 cells (, ). In both cases, Claspin expression levels were not obviously affected (, ). Combined inhibition of ZEB1 and TopBP1 demonstrated that ZEB1 inhibition facilitated Chk1 phosphorylation by enhancing TopBP1 expression (). When ZEB1 siRNA-treated PANC-1 and MGC-803 cells were treated with VE-821, we found that the ZEB1 inhibition-induced increase in Chk1 phosphorylation was almost completely abolished in both cell lines (). Finally, the effects of ZEB1 inhibition on the cell cycle were determined by flow cytometry. ZEB1 knockdown using siRNA resulted in a greater percentage of cells in the S phase of the cell cycle compared with scrambled siRNA treatment (39.1 ± 2.8% vs 27.2 ± 1.8%, p = 0.001) (). These results demonstrate for the first time that ZEB1 inhibition promotes Chk1 phosphorylation by enhancing TopBP1 expression, and induces S-phase arrest.
Figure 4. ZEB1 inhibition promoted Chk1 phosphorylation via increasing TopBP1 expression and induces S-phase arrest. (A) PANC-1 and MGC-803 cells were transfected with Scrambled Control siRNA or two pairs of ZEB1 siRNA, the expression of ZEB1, p-Chk1, total Chk1 and ATR was determined by Western Blotting. (B) MGC-803 cells were transfected with Scrambled Control siRNA or ZEB1 siRNA, then exposed to 1mM HU for 2 h. The expression of ZEB1, p-Chk1and total Chk1 was detected by Western Blotting. (C) HCT-116 cells were transiently transfected with pcDNA3.1/ZEB1-Flag plasmid or pcDNA3.1 empty control, then exposed to 1mM HU for 2 h. The expression of ZEB1, p-Chk1 and total Chk1 was detected by Western Blotting. (D) PANC-1 and MGC-803 cells were transfected with Scrambled Control siRNA or two pairs of ZEB1 siRNA, the expression of TopBP1 and Claspin was determined by Western Blotting. (E) HCT-116 cells were transiently transfected with pcDNA3.1/ZEB1-Flag plasmid or pcDNA3.1 empty control, the expression of TopBP1 and Claspin was determined by Western Blotting. (F) MGC-803 cells were transfected with Scrambled Control siRNA, ZEB1 siRNA, TopBP1 siRNA or combined ZEB1 siRNA and TopBP1 siRNA. The expression of ZEB1, TopBP1, p-Chk1and total Chk1 was determined by Western Blotting. (G) PANC-1 and MGC-803 cells were transfected with Scrambled Control siRNA or ZEB1 siRNA, then exposed to 5 μM VE-821 for 24 h. The expression of p-Chk1 and total Chk1 was determined by Western Blotting. (H) MGC-803 cells were transfected with Scrambled Control siRNA or ZEB1 siRNA, then the proportion of cell cycle phase was examined by flow cytometric assay. *p <0.05 vs. scrambled control.
ZEB1 inhibition combined with VE-821 enhanced DNA damage accumulation
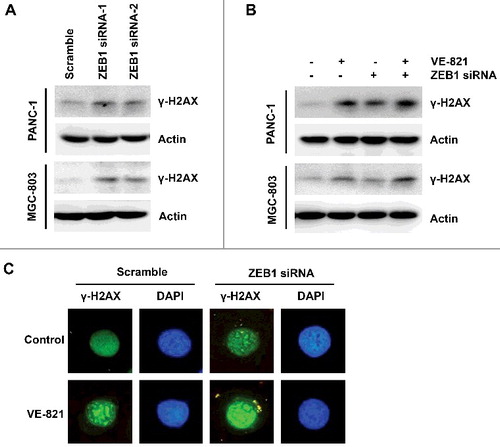
Because treatment with an ATR inhibitor leads to DNA damage accumulation, we conducted western blotting and immunofluorescence to monitor changes in the DNA damage marker γ-H2AX when cells were treated with VE-821 combined with ZEB1 inhibition. ZEB1 downregulation by siRNA increased DNA damage, as verified by γ-H2AX expression and γ-H2AX-positive foci in both PANC-1 and MGC-803 cells (, ). Furthermore, DNA damage accumulation was further enhanced when VE-821 treatment was combined with ZEB1 inhibition, as demonstrated by a further increase in the γ-H2AX level and number of γ-H2AX-positive foci (, ). These results demonstrate that VE-821 combined with ZEB1 inhibition led to further accumulation of DNA damage in cells undergoing VE-821-induced EMT.
Figure 5. ZEB1 inhibition enhanced DNA damage to VE-821. (A) PANC-1 and MGC-803 cells were transfected with Scrambled Control siRNA or two pairs of ZEB1 siRNA, the expression of γ-H2AX was determined by Western Blotting. (B) PANC-1 and MGC-803 cells were transfected with Scrambled Control siRNA or ZEB1 siRNA, then exposed to 5 μM VE-821 for 24 h. The expression of γ-H2AX was determined by Western Blotting. (C) MGC-803 cells were transfected with Scrambled Control siRNA or ZEB1 siRNA, then exposed to 5 μM VE-821 for 24 h. Positive foci of γ-H2AX was performed by immunofluorescence assay.
Discussion
Inhibition of ATR and Chk1 is an essential treatment strategy in ATM- and p53-deficient cancers, because such defects drive cancer cells reliant on the ATR/Chk1 pathway for DNA damage repair. Although inhibition of ATR increases sensitivity to radiation and chemotherapy, application of ATR inhibitors alone has achieved limited clinical benefits. Therefore, exploring the factors regulating the sensitivity of cancer cells to ATR inhibitors is of clinical significance. In this study, we found that the ATR inhibitor VE-821 induced EMT accompanied by ZEB1 upregulation and activation of proliferative signaling in PANC-1 and MGC-803 cells. Inhibition of ZEB1 abrogated VE-821-induced EMT and enhanced the sensitivity of the cells to VE-821. Furthermore, ZEB1 inhibition induced Chk1 phosphorylation and S-phase arrest.
We previously demonstrated that EMT promoted migration and metastasis in gastric cancer cells [Citation18]. Moreover, EMT is associated with resistance to chemotherapy and radiotherapy in various cancers. However, whether EMT confers sensitivity to ATR inhibitors was unknown. When we applied VE-821 to various cancer cell lines, we found that the different cell types underwent different morphological changes. VE-821 induced EMT in PANC-1 and MGC-803 cells, whereas it inhibited proliferation without inducing EMT in HCT-116 and NCI-N87 cells, indicating that EMT mediated the sensitivity of cells to VE-821. Several transcription factors are involved in EMT. Among the common EMT-associated transcription factors, only ZEB1 upregulation was associated with VE-821-induced EMT. To confirm the role of ZEB1 in modulating the sensitivity of cells to VE-821, we used siRNA to knock down ZEB1 expression. Further experiments demonstrated that ZEB1 inhibition enhanced the sensitivity of cells to VE-821 through suppression of EMT and proliferative signaling. Generally, ZEB1 is located downstream of ERK/AKT-miR-200c-ZEB1 axis [Citation19]. However, few studies have reported the regulation of AKT by ZEB1. Guo Y's study indicated that ZEB1 knockdown decreased AKT phosphorylation [Citation20], which was consistent with our study, but the mechanism was not explored. These results indicate that EMT may limit the benefits of ATR inhibitors in clinical practice, and that reversing EMT, especially by inhibiting ZEB1, may provide an alternative approach for treating patients resistant to ATR inhibitors.
Two studies investigating the correlation between the ATR/Chk1 pathway and EMT have suggested that ATM/ATR is related to the EMT-inducing transcription factors ZEB1 and ZEB2 [Citation21,22]. In response to DNA damage, ATM phosphorylated and stabilized ZEB1, and ZEB1 in turn promoted resistance to radiation by stabilizing Chk1 [Citation21]. Another study indicated that ZEB2 suppressed phosphorylation of ATM/ATR substrates but had no effect on the level of DNA damage [Citation22]. Here, we explored the modulation of Chk1 by ZEB1. Unlike in the previous study, we found that ZEB1 inhibition promoted Chk1 phosphorylation and S-phase arrest in PANC-1 cells and MGC-803 cells, and ZEB1 overexpression attenuated HU-stimulated Chk1 phosphorylation in HCT-116 cells. Furthermore, we demonstrated that ZEB1 inhibition increased expression of TopBP1, which is the main ATR adaptor protein that enhances Chk1 phosphorylation. To further define the relationship between ZEB1 expression and Chk1 expression in colon cancer, we examined gene expression data from the TCGA colon adenocarcinoma (COAD) consortium. Spearman's correlation analysis indicated no significant relationship between ZEB1 mRNA expression and Chk1 mRNA expression (γ = 0.031, p = 0.503) (Supplemental Fig. 3). Such diverse results suggest that the regulation of Chk1 by ZEB1 is complex, and varies between cell lines or according to other undefined factors. The detailed mechanisms are worthy of exploration.
Because ATR is an essential component of homologous recombination (HR) repair, ATR inhibitors reduce HR repair capacity and induce DNA damage accumulation [Citation1]. Previous studies have indicated that ZEB1 is required for HR-mediated DNA damage repair [Citation21]. Therefore, we explored changes in the foci of the DNA damage marker γ-H2AX induced by VE-821 treatment combined with ZEB1 inhibition. VE-821 treatment or ZEB1 inhibition alone enhanced DNA damage, and the two combined further augmented DNA damage accumulation, assessed by measuring γ-H2AX foci. This suggests another mechanism for ZEB1 inhibition-induced ATR inhibitor sensitization.
Taken together, our findings demonstrate for the first time that EMT can limit the effectiveness of an ATR inhibitor, associated with a reduction in inhibition of migration and activation of proliferative signaling. Inhibiting ZEB1 not only reversed VE-821 -induced EMT and enhanced inhibition of migration, but also suppressed activation of proliferative signaling. Additionally, ZEB1 inhibition enhanced Chk1 phosphorylation and DNA damage. These effects contributed to the ATR inhibitor-sensitizing effect of ZEB1 inhibition, suggesting that targeting ZEB1 might enhance the efficacy of ATR inhibitors or reverse resistance to these drugs in cells exhibiting EMT.
Materials and methods
Cell culture
Human pancreatic cancer cell line PANC-1, gastric cancer cells MGC-803 and NCI-N87, colon cancer cells HCT-116 was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), which was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Gaithersburg, MD, USA) with 10% fetal bovine serum (FBS).
Reagents and antibodies
The ATR inhibitor VE-821 was purchased from Selleck chemicals (Houston, Texas, USA). Hydroxyurea (HU) was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Antibodies to E-cadherin, Vimentin, phospho-Chk1 (S345), phospho-AKT (S473), total AKT, phospho-ERK1/ERK2 (Thr202/Tyr204), γ-H2AX, FLAG M2 were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies to total ATR, Chk1, TopBP1 and Claspin were obtained from Bethyl Laboratories (Montgomery, TX, USA). Antibodies to Twist2, Slug, Snail was purchased from Abcam (Cambridge, UK). Anti-Actin, Cdc25A, total ERK, ZEB1, ZEB2, secondary goat anti-mouse and goat anti-rabbit antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell viability assay
Cancer cells were plated in 96-well plates in triplicate at 5000 cells/well. The next day, the cells were exposed to increasing concentrations of VE-821 for 48 h. The effect of cell proliferation was measured using a 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.
Western blotting
Western blotting was performed as described in our previous study [Citation23]. Briefly, the cells were lysed in lysis buffer (1% Triton X-100, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 2 µg/ml aprotinin) and quantified using the BCA method. Following separation by 8% SDS-PAGE, the samples were transferred to a nitrocellulose membrane. After blocking with 5% skim milk in tris-buffered saline Tween-20 (TBST) buffer, antibodies were added and incubated overnight at 4 ˚C. Following secondary antibodies incubation and TBST buffer washes, the protein bands were detected with enhanced chemiluminescence reagent using the Electrophoresis Gel Imaging Analysis System.
Migration assay
Migration assays were carried out in the 24-well Boyden chamber (8-μm pore size; Corning, USA). After pretreatment with ZEB1 siRNA or exposed to VE-821 for 24 h, 2 × 104 cells were loaded into the upper chamber with 200 μl serum-free RPMI 1640 medium. The lower chambers contained 500 µl of RPMI 1640 with 2.5% FBS. Then cultured for 24h, the migrated cells onto the outer side of the membrane were stained with Trypan Blue and five random fields per well were captured and counted.
Small interfering RNA transfections
The siRNA sequences were from View solid biotechnology co., LTD (Beijing, China). Two different pairs of ZEB1 siRNA were 5'-GGCGGUAGAUGGUAAUGUAtt-3' and 5'-GUCGCUACAAACAGUUGUAtt-3'. TopBP1 siRNA was 5'- GCUCUGUAAUAGUCGACUAtt-3'. The siRNAs were transfected with Lipofectamine 2000 per the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA).
Plasmid construction
PcDNA3.1/ZEB1-Flag plasmid was designed and synthesized from Obio Technology Co., Ltd. (Shanghai, China). HCT-116 cells were transfected with 1.5 μg pcDNA3.1/ZEB1-Flag or pcDNA3.1 empty vector using Lipofectamine 2000 according to the manufacturer's instructions.
Immunofluorescence imaging
The cells were seeded in Lab-Tek chamber slides (Nunc S/A, Polylabo, Strasbourg, France). After treated with VE-821 or ZEB1 siRNA for the indicated time, the cells were fixed in 3.3% paraformal-dehyde for 15 min and permeabilized with 0.2% Triton X-100 for 5 min. Then the cells were blocked with 5% bovine serum albumin (BSA) for 1 hour and incubated with anti-Vimentin or anti-γ-H2AX antibody overnight at 4°C. Then the Alexa Fluor 488-conjugated goat anti-rabbit IgG was added in blocking solution for 1 hour at room temperature in the dark and 4′6′-diamidino-2- phenylindole (DAPI) was used to stain nuclei for 5 min. Finally, the cells were mounted with the Slow Fade Antifade Kit and visualized by fluorescence microscopy (BX61, Olympus, Japan).
Flow cytometry assay
The cells were collected and washed twice with PBS. After fixing in 70 % ethanol for 12 h, the samples were washed with PBS and incubated with 20 μg/ml RNase A for 30 min and 10μg/ml propidium iodide (PI) for 30 min in the dark. Finally, the samples were evaluated by flow cytometry (BD Accuri C6; BD Biosciences, San Jose, CA, USA), and analyzed with WinMDI software version 2.9 (The Scripps Research Institute, La Jolla, CA, USA).
Statistical analysis
The differences of the results between two groups were evaluated by Student's t-test. p-value of 0.05 or less was considered statistically significant.
Abbreviations
| ATM, | = | ataxia-telangiectasia mutated |
| ATR, | = | ATM and Rad3-related |
| DSBs, | = | double-stranded DNA breaks |
| Chk1, | = | checkpoint kinase 1 |
| HR, | = | homologous recombination |
| EMT, | = | epithelial-to-mesenchymal transition |
| ZEB1, | = | zinc finger E-box binding homeobox 1 |
| DDR, | = | DNA damage repair |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the National Key Research and Development Program of China (No.2017YFC1308900), National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2017ZX09304025), National Natural Science Foundation of China (No.81401938, No.81572374), Science and Technology Plan Project of Liaoning Province (No.2014226033), The general project of Liaoning province department of education (No.LS201613), Project for clinical ability construction of Chinese medicine, Science and Technology Plan Project of Liaoning Province (2016007010).
Supplementary.rar
Download (2.3 MB)Additional information
Funding
Related Research Data
References
- Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet. 2016;50:155–173. doi:10.1146/annurev-genet-121415-121658. PMID:27617969
- Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21:4780–4785. doi:10.1158/1078-0432.CCR-15-0479. PMID:26362996
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi:10.1101/gad.914401. PMID:11544175
- Zou L. Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007;21:879–885. doi:10.1101/gad.1550307. PMID:17437994
- Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–430. doi:10.1038/nchembio.573. PMID:21490603
- Kim HJ, Min A, Im SA, et al. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017;140:109–119.
- Prevo R, Fokas E, Reaper PM, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–1081. doi:10.4161/cbt.21093. PMID:22825331
- Fokas E, Prevo R, Pollard JR, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi:10.1038/cddis.2012.181. PMID:23222511
- Jossé R, Martin SE, Guha R, et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014;74:6968–6979. doi:10.1158/0008-5472.CAN-13-3369. PMID:25269479
- Min A, Im SA, Jang H, et al. AZD6738, a novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol Cancer Ther. 2017;16:566–577. doi:10.1158/1535-7163.MCT-16-0378. PMID:28138034
- Krajewska M, Fehrmann RS, Schoonen PM, et al. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene. 2015;34:3474–3481. doi:10.1038/onc.2014.276. PMID:25174396
- Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–10711. doi:10.18632/oncotarget.4037. PMID:25986923
- Chang L, Graham PH, Hao J, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi:10.1038/cddis.2013.407. PMID:24157869
- Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–487. doi:10.1080/15384101.2015.1006048. PMID:25607528
- Wang C, Jin H, Wang N, et al. Gas6/Axl Axis contributes to chemoresistance and metastasis in breast cancer through Akt/GSK-3β/β-catenin signaling. Theranostics. 2016;6:1205–1219. doi:10.7150/thno.15083. PMID:27279912
- Zou J, Liu L, Wang Q, et al. Downregulation of miR-429 contributes to the development of drug resistance in epithelial ovarian cancer by targeting ZEB1. Am J Transl Res. 2017;9:1357–1368. PMID:28386361
- Zhou G, Zhang F, Guo Y, et al. miR-200c enhances sensitivity of drug-resistant non-small cell lung cancer to gefitinib by suppression of PI3K/Akt signaling pathway and inhibites cell migration via targeting ZEB1. Biomed Pharmacother. 2017;85:113–119. doi:10.1016/j.biopha.2016.11.100. PMID:27930974
- Li H, Xu L, Li C, et al. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol Cancer. 2014;13:136. doi:10.1186/1476-4598-13-136. PMID:24885194
- Xu L, Zhang Y, Qu X, E3 Ubiquitin ligase Cbl-b prevents tumor metastasis by maintaining the epithelial phenotype in multiple drug-resistant gastric and breast cancer cells. Neoplasia. 2017;19:374–382. doi:10.1016/j.neo.2017.01.011. PMID:28334634
- Guo Y, Lang X, Lu Z, et al. MiR-10b directly targets ZEB1 and PIK3CA to curb adenomyotic epithelial cell invasiveness via upregulation of E-Cadherin and inhibition of Akt phosphorylation. Cell Physiol Biochem. 2015;35:2169–2180. doi:10.1159/000374022. PMID:25896413
- Zhang P, Wei Y, Wang L, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864–875. doi:10.1038/ncb3013. PMID:25086746
- Sayan AE, Griffiths TR, Pal R, et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci U S A. 2009;106:14884–14889. doi:10.1073/pnas.0902042106. PMID:19706487
- Song N, Liu S, Zhang J, et al. Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int J Mol Sci. 2014;15:5838–5851. doi:10.3390/ijms15045838. PMID:24714091