
ABSTRACT
Metabolic reprogramming is a hallmark of cancer cells, but the mechanisms are not well understood. The mammalian target of rapamycin complex 2 (mTORC2) controls cell growth and proliferation and plays a critical role in metabolic reprogramming in glioma. mTORC2 regulates cellular processes such as cell survival, metabolism, and proliferation by phosphorylation of AGC kinases. Components of mTORC2 are shown to localize to the nucleus, but whether mTORC2 modulates epigenetic modifications to regulate gene expression is not known. Here, we identified histone H3 lysine 56 acetylation (H3K56Ac) is regulated by mTORC2 and show that global H3K56Ac levels were downregulated on mTORC2 knockdown but not on mTORC1 knockdown. mTORC2 promotes H3K56Ac in a tuberous sclerosis complex 1/2 (TSC1/2) mediated signaling pathway. We show that knockdown of sirtuin6 (SIRT6) prevented H3K56 deacetylation in mTORC2 depleted cells. Using glioma model consisting of U87EGFRvIII cells, we established that mTORC2 promotes H3K56Ac in glioma. Finally, we show that mTORC2 regulates the expression of glycolytic genes by regulating H3K56Ac levels at the promoters of these genes in glioma cells and depletion of mTOR leads to increased recruitment of SIRT6 to these promoters. Collectively, these results identify mTORC2 signaling pathway positively promotes H3K56Ac through which it may mediate metabolic reprogramming in glioma.
Introduction
The signaling mechanisms by which environmental factors affect epigenetic processes and chromatin structure to regulate gene transcription are poorly understood. The mammalian target of rapamycin (mTOR) pathway responds to intracellular and extracellular signals and functions as a central regulator of cell growth, proliferation, and metabolism [Citation1]. mTOR is a serine/threonine kinase which interacts with several proteins and forms two discrete complexes, mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2). The mTORC1 and mTORC2 complexes share catalytic subunits mTOR, MLST8, and DEPTOR. Raptor and PRAS40 are the specific subunits of mTORC1, whereas RICTOR, mSIN1, and Protor1/2 are the specific subunits of mTORC2 complex [Citation1]. The mTORC1 integrates signaling from growth factors, energy status, and nutrients to promote cell growth and is acutely sensitive to rapamycin [Citation2]. The mTORC2 signaling is responsive to growth factors but insensitive to nutrients [Citation3–5]. TSC1/2 complex senses signals from upstream signaling pathways and acts as a key regulator of mTOR signalling. TSC complex inhibits mTORC1 by acting as Rheb GTPase activating protein (GAP). mTORC2 requires TSC complex for its activity [Citation6]. Till now, the well characterized direct substrates of mTORC2 are AGC subfamily of kinases including Akt, serum and glucocorticoid-induced protein kinase 1 (SGK1), and protein kinase C-α (PKC-α). It regulates cellular processes such as cell survival, metabolism, and proliferation by phosphorylation of Akt, SGK1 and PKCα proteins [Citation7]. Whether mTORC2 targets any other class of proteins is not known. Components of mTORC1 and mTORC2 complexes are shown to be present in nucleus [Citation8,Citation9]. In yeast, TORC1 localizes to the nucleus and regulates gene transcription, cell cycle, and epigenetic modifications [Citation10]. In mammalian cells, mTOR binds to the promoters of Pol I and Pol III-transcribed genes to coordinate transcription [Citation11–13]. Whether the mTORC2 complex has any such functions in the nucleus or its role in regulating transcription has not been studied. Histone H3 lysine 56 acetylation (H3K56Ac) is a core histone modification conserved from yeast to humans. H3K56Ac is localized on thousands of promoters in embryonic stem cells and linked to core transcriptional network [Citation14]. Acetylation of H3K56 on the promoters of genes induces their expression [Citation15,Citation16]. H3K56Ac is altered in response to DNA damage and is required for DNA damage signaling and recovery [Citation17–19]. Sirtuins are the homologs of Sir2 proteins in yeast [Citation20]. SIRT6 is localized to the nucleus and is a chromatin-bound protein [Citation21]. Deacetylation of H3K56 by SIRT6 has important functions in transcription, telomere maintenance, DNA damage, glucose metabolism, and cellular transformation [Citation22,Citation23]. A recent report suggested that loss of SIRT6 sumoylation affects SIRT6 deacetylase activity specifically towards H3K56, resulting in loss of tumor suppressor function [Citation24].
Crosstalk between TOR and sirtuin pathways has been reported. In yeast, TORC1 acts as a negative regulator of Sir2 by suppressing the association of Sir2 with rDNA. Inhibition of TORC1 resulted in deacetylation of histones by Sir2 on rDNA [Citation25]. In a recent study, it has been shown that TORC1 signaling positively regulates H3K56Ac by negatively regulating the HST3 and HST4 proteins [Citation26]. In mammals, mTORC1 and mTORC2 complexes induce metabolic reprogramming in tumors by upregulating the expression of metabolic genes [Citation27,Citation28]. On the other hand, SIRT6 is a tumor suppressor and deacetylates H3K56 on the promoters of ribosomal and glycolytic genes leading to their reduced expression [Citation23,Citation29]. The inverse relationship between the roles of SIRT6 and mTOR in the regulation of glycolytic metabolism suggests a functional interrelationship between these two proteins.
Here, we report for the first time that H3K56Ac is regulated by mTORC2 and not mTORC1 in mammalian cells. mTORC2 regulates H3K56Ac independent of AKT signaling. We also show that TSC1/2 complex functions upstream of mTORC2 to regulate H3K56Ac and SIRT6 deacetylates H3K56 in the absence of mTORC2. Further, in glioma model of U87EGFRvIII cells, we report that mTORC2 promotes H3K56Ac at the promoters of glycolytic genes by negatively regulating the recruitment of SIRT6 to regulate their transcription.
Results
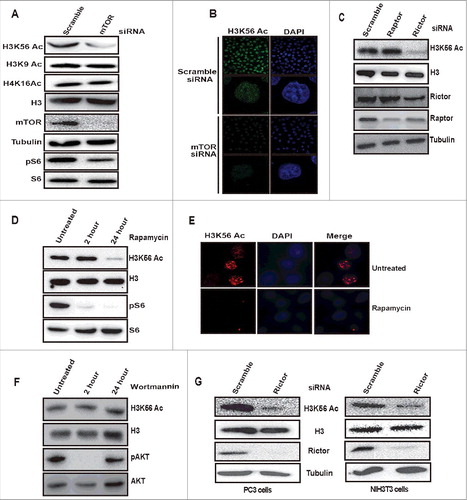
mTORC2 regulates Histone H3 lysine 56 acetylation
In response to changes in the extracellular and intracellular environment, chromatin modification patterns are altered to regulate gene expression [Citation30]. Previously, it has been reported that histone acetylation is controlled by universally conserved environmental regulated TORC1 signaling pathway in yeast [Citation10,Citation26,Citation31]. However, the connection between mTOR signaling and histone acetylation has not been studied much in the mammalian system. To investigate the role of mTOR pathway in the regulation of histone acetylation in the mammalian system, we depleted mTOR in HeLa cells by siRNA and examined the acetylation status of histone H3 and H4 modifications such as H3K56Ac, H3K9Ac, and H4K16Ac by immunoblotting with residue specific antibodies. Our results showed a reduction of H3K56Ac in mTOR depleted cells (). We did not find any change in H3K9Ac and H4K16Ac levels on knockdown of mTOR, suggesting mTOR signaling positively regulates H3K56Ac (). Immunofluorescence analysis further confirmed the reduced levels of H3K56Ac in mTOR depleted HeLa cells (). Downregulation of phosphorylation on S6 (ser240/244) suggested the downregulation of mTOR signaling in knockdown lysates (). mTOR kinase exists in two complexes, mTORC1 and mTORC2. The mTORC1 consists of mTOR, Raptor, and mLST8 and mTORC2 consists of mTOR, rictor, and mSIN1. Raptor and Rictor are the essential specific core components of mTORC1 and mTORC2 respectively [Citation2]. In order to determine which mTOR complex regulates H3K56Ac, we have knocked down raptor and rictor by siRNA in HeLa cells. Depletion of rictor but not raptor resulted in decreased H3K56Ac levels (), indicating that mTORC2 (mTOR/rictor) regulates H3K56Ac but not mTORC1 (mTOR/Raptor). The levels of H3K9 and H4K16 acetylation were unaltered in rictor knockdown cell lysates (Supplementary Figure S1). Notably, knockdown of any of the mTOR complex proteins has no effect on cell proliferation in HeLa cells (Supplementary Figure S2), indicating that downregulation of H3K56Ac on mTOR complex depletion is not a result of changes in cell proliferation. mTORC1 is acutely sensitive to rapamycin [Citation1,Citation2], whereas prolonged treatment with rapamycin affects mTORC2 assembly and activity [Citation9,Citation32]. To determine whether prolonged rapamycin treatment affects H3K56Ac, HeLa cells were treated with 100 nM rapamycin for 2 h and 24 h. Our western blot result showed that H3K56Ac remained unchanged at 2 h but decreased in cells treated for 24 h (). Immunofluorescence analysis of H3K56Ac on rapamycin treatment for 24 h also showed decreased acetylation on H3K56 (). Rapamycin acutely inhibits mTORC1 and has anti-proliferative effects [Citation1]. Treatment of HeLa cells with rapamycin for 24 h has no effect on cell proliferation (Supplementary Figure S3), suggesting that the decreased H3K56Ac on rapamycin treatment is not due to cell proliferation defects. mTORC2 phosphorylates Akt at serine473 and plays a key role in multiple cellular signaling pathways [Citation7]. Activated Akt (pAktSer473) has been shown to promote global histone acetylation in cancer cells [Citation33]. To investigate whether the decreased H3K56Ac in rictor knockdown is due to downregulated Akt phosphorylation, we treated HeLa cells with wortmannin (PI3kinase inhibitor) and observed unaltered H3K56Ac levels (), suggesting that mTORC2 complex regulates H3K56Ac independent of Akt signaling. Depletion of rictor in PC3 and NIH3T3 cell lines also resulted in decreased H3K56Ac (), suggesting that mTORC2 mediated regulation of H3K56Ac is conserved among different cell lines. Overall, these results demonstrate that mTORC2 signaling positively regulates H3K56Ac.
Figure 1. mTORC2 regulates H3K56 acetylation. (A) mTOR signaling positively regulates global H3K56Ac levels but not H3K9Ac and H4K16Ac. HeLa cells were transfected with scramble or mTOR siRNA using lipofectamine reagent. After 48 h of transfection, cells were harvested, whole cell lysates were prepared and resolved on SDS-PAGE and levels of indicated proteins were analyzed by Western blot. (B) HeLa cells were transfected with scramble or mTOR siRNA. After 48 h of transfection, cells were fixed with paraformaldehyde. Immunofluorescence was performed using anti-H3K56Ac antibodies. Images shown are from a representative of multiple experiments. (C) Rictor but not raptor knockdown downregulates H3K56Ac. HeLa cells were transfected with raptor or rictor siRNA. After 48 h cells were harvested and whole cell lysates were prepared. Lysates were resolved on SDS-PAGE and the levels of H3K56Ac was analyzed by Western blot. (D) Prolonged treatment with rapamycin downregulates H3K56Ac. HeLa cells were treated with 100nM rapamycin for 2 and 24 h, whole cell lysates were prepared and equal quantities of cell lysates were resolved on SDS-PAGE and the levels of H3K56Ac was analysed by Western blot. (E) HeLa cells were treated with rapamycin (100nM) for 24 h. Cells without treatment were taken as controls. After 24 h treatment, cells were fixed with paraformaldehyde and immunofluorescence was performed with anti-H3K56Ac antibodies. (F) H3K56Ac levels were unchanged on Wortmannin treatment. HeLa cells were kept untreated or treated with wortmannin (100nM) for 2 h or 24 h and harvested. Whole cell lysates were resolved on SDS-PAGE and the levels of indicated proteins were analyzed by Western blot. (G) mTORC2 mediated regulation of H3K56Ac is conserved in different cell lines. PC3 and NIH3T3 cells were transfected with scramble or rictor siRNA. After 48 h of transfection, cells were harvested and whole cell lysates were prepared, resolved on SDS-PAGE. Levels of H3K56Ac were analyzed by western blot. Total H3 and Tubulin were used as loading controls.
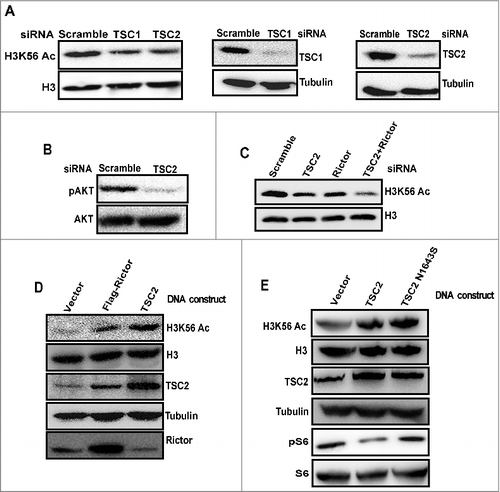
TSC complex is required to maintain global H3K56 acetylation levels
In response to environmental changes such as growth factors, cellular nutrients, and energy status, mTOR is controlled by several upstream regulators [Citation1,Citation2]. TSC (TSC1/2) inhibits mTORC1 and is required for mTORC2 activity [Citation6,Citation34,Citation35]. To investigate whether signaling from TSC complex affect H3K56Ac, we knocked down TSC1 and TSC2 proteins by siRNA. Immunoblot analysis indicated reduced H3K56Ac in TSC1 and TSC2 depleted cells (), suggesting that TSC1/TSC2 complex is required for acetylation of H3K56 upstream of the mTORC2 complex. Phosphorylation of AKT on ser473 was decreased in TSC2 knockdown lysates indicating downregulated mTORC2 activity (). Further, knockdown of both rictor and TSC2 resulted in a pronounced decrease in H3K56Ac levels than individual protein knockdowns (), suggesting that TSC2 might regulate H3K56Ac through mTORC2 dependent and independent pathway. Overexpression of rictor and TSC2 plasmid constructs in HeLa cells resulted in increased H3K56Ac in TSC2 and rictor overexpressing cells (). TSC acts predominantly as a negative regulator of mTORC1 by acting as Rheb GAP [Citation1]. Also, mTORC1 has been reported to negatively regulate mTORC2 [Citation36]. To deconvolute the regulation of mTORC2 by mTORC1 and mTORC1 by TSC, we have overexpressed TSC2N1643S (GAP mutant, which cannot suppress Rheb activity) in HeLa cells. Our result showed increased acetylation on H3K56 in TSC2N1643S expressing HeLa cells (), suggesting that TSC regulates H3K56Ac independent of Rheb-GTPase activity. Overall, these results implicate that TSC1/2 complex positively regulates H3K56Ac through mTORC2 dependent and independent pathway.
Figure 2. TSC complex functions upstream of mTORC2 to regulate H3K56 acetylation. (A) TSC complex positively regulates H3K56Ac. HeLa cells were transfected with scramble, TSC1 or TSC2 siRNA. After 48 h of transfection, cells were harvested. Whole cell lysates were prepared and resolved on SDS-PAGE and levels of H3K56Ac were analyzed by Western blot. (B) mTORC2 mediated AKTser473 phosphorylation is downregulated in TSC2 depleted cells. HeLa cells were transfected with TSC2 siRNA. After 48 h, cells were harvested and whole cell lysates were analysed for Aktser473 phosphorylation by Western blot. AKT was probed as a loading control. (C) TSC2 regulate H3K56Ac by both mTORC2 dependent and independent pathways. HeLa cells were transfected with either scramble, TSC2 or rictor siRNA alone or transfected with both TSC2 and rictor siRNA. After 48 h of transfection, cells were harvested and whole cell lysates were loaded on SDS-PAGE and analyzed for H3K56Ac by Western blot. (D) Overexpression of rictor and TSC2 proteins induce H3K56Ac. HeLa cells were transfected with flag-rictor or pcDNA3.1-TSC2 DNA constructs for 24 h. Whole cell lysates were prepared and resolved on SDS-PAGE. Levels of H3K56Ac was analysed by Western blot. H3 and Tubulin were probed as loading controls. (E) Overexpression of TSC2N1643S (GAP mutant) induce H3K56Ac. HeLa cells were transfected with TSC2 or TSC2N1643S DNA constructs for 24h. Whole cell lysates were analyzed for the levels of H3K56Ac by Western blot.
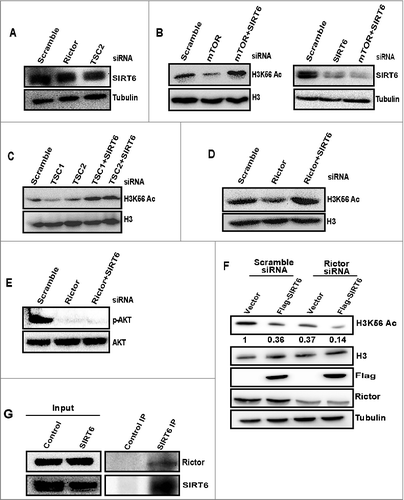
SIRT6 deacetylates H3K56 in the absence of mTORC2
In S. cerevisiae H3K56 is deacetylated by Sir2 and its homologs, Hst3 and Hst4 [Citation37,38]. It has been reported that deletion of Hst3 or Hst4 proteins rescues H3K56Ac in TORC1 mutants [Citation26]. In mammals, there are seven Sirtuins (SIRT1-7). Among the seven sirtuins SIRT1, SIRT2, and SIRT6 have been reported to deacetylate H3K56Ac in mammals [Citation17,39,40]. We were interested in studying the regulation of H3K56Ac by SIRT6, as it is predominantly a chromatin-bound protein and deacetylation of H3K56Ac by SIRT6 has wide roles in chromatin regulation [Citation22]. Hence, we investigated the role of SIRT6 in mTORC2 mediated regulation of H3K56Ac. Initially, we checked whether SIRT6 protein levels are altered in mTOR, rictor, and TSC2 depleted cells. Our western blot analysis showed that SIRT6 levels were unchanged in mTOR, rictor, and TSC2 knockdown cell lysates (), indicating that decreased acetylation is not due to upregulated SIRT6 expression. Next, we wanted to determine whether deletion of SIRT6 can rescue H3K56Ac levels in the absence of mTOR. In that context, we have transfected cells with SIRT6 siRNA along with mTOR, rictor, and TSC2. Deacetylation of H3K56 has rescued in all co-depletions, indicating that SIRT6 deacetylates H3K56 in the absence of mTORC2 signaling (, Supplementary Figure S4). Levels of pAktser473 were not rescued in rictor and SIRT6 double knockdown cells (), suggesting that SIRT6 depletion rescues H3K56Ac independent of AKT signaling. SIRT6 binds tightly to chromatin and deacetylates H3K56 and H3K9 [Citation21,41]. Localization of SIRT6 on chromatin is dynamic and DNA damage triggers its increased association on chromatin [Citation42,43]. Hence, to investigate whether mTOR depletion affects localization of SIRT6 on chromatin, we fractionated chromatin-bound proteins as previously described [Citation42–44] to examine the localization of SIRT6 on chromatin in mTOR depleted cells. Our Western blot data showed no obvious change in the localization of SIRT6 on chromatin in the absence of mTOR (Supplementary Figure S5), suggesting that mTOR signaling does not affect global SIRT6 localization on chromatin. SIRT6 has a poor deacetylase activity in vitro [Citation40,45–47]. This might be due lack of post-translational modifications or cofactors needed for its activity in in vitro studies. To test whether rictor modulates the activity of SIRT6, we performed an in vivo deacetylase activity of SIRT6 by overexpressing SIRT6 in rictor depleted HeLa cells and assessed for deacetylase activity of SIRT6 by checking levels of H3K56Ac. Our results revealed that the deacetylase activity of SIRT6 on H3K56 increased significantly in rictor depleted cells overexpressing SIRT6 than control cells overexpressing SIRT6 (lanes 2 and 4) (), indicating that SIRT6 activity towards H3K56Ac is increased in the absence of mTORC2. mTORC1 and mTORC2 components are reported to be present abundantly in cytoplasm and nucleus [Citation8,9]. It has been reported that mTOR/Rictor complex is abundant in the nucleus [Citation9]. To check the interaction between SIRT6 and rictor, whole cell lysates from HeLa cells were subjected to co-immunoprecipitation with SIRT6 antibody and immunoblotted with rictor antibody. Our western blot data showed an interaction between rictor and SIRT6 (). Overall, these results revealed that SIRT6 mediates the deacetylation of H3K56 in the absence of mTORC2 signaling and deacetylase activity of SIRT6 toward H3K56Ac is increased in the absence of mTORC2.
Figure 3. SIRT6 deacetylates H3K56Ac in the absence of mTORC2. (A) Disruption of mTORC2 signaling does not alter SIRT6 expression. HeLa cells were transfected with scramble, rictor, or TSC2 siRNA. After 48 h of transfection, cells were harvested and whole cell lysates were resolved on SDS-PAGE. Levels of SIRT6 were analyzed by Western blot. (B) SIRT6 deacetylates H3K56 in the absence of mTOR. HeLa cells were transfected with scramble, mTOR or in a combination of mTOR and SIRT6 siRNA. After 72 h of transfection, whole cell lysates were analyzed for H3K56Ac levels by Western blot. (C) SIRT6 deacetylates H3K56 in the absence of TSC complex. HeLa cells were transfected with either scramble, TSC1, TSC2 siRNA or in combination with SIRT6 siRNA. After 48 h of transfection, whole cell lysates were analyzed for H3K56Ac levels by Western blot. (D) The decrease in H3K56Ac resulting from rictor depletion is recovered by SIRT6 knockdown. HeLa cells were transfected with either scramble, rictor siRNA or in combination with SIRT6 siRNA. After 48 h of transfection, whole cell lysates were analysed for H3K56Ac levels by Western blot. (E) Decrease in AKT S473 phosphorylation resulting from rictor depletion cannot be rescued by SIRT6 knockdown. HeLa cells were transfected with scramble or rictor siRNA alone or transfected with rictor and SIRT6 siRNA. After 48 h of transfection, whole cell lysates were analyzed for Aktser473 phosphorylation and AKT levels by Western blot. (F) mTORC2 negatively regulate deacetylase activity of SIRT6. HeLa cells were transfected with scramble or rictor siRNA for 48 h and then cells were transfected with the FLAG-SIRT6 plasmid for 24 hours. Whole cell lysates were resolved on SDS-PAGE and analyzed for indicated proteins by Western blot. (G) SIRT6 interacts with rictor. Endogenous SIRT6 was co-immunoprecipitated from HeLa cells with SIRT6 specific antibody and lysates were analyzed for the presence of rictor by Western blot.
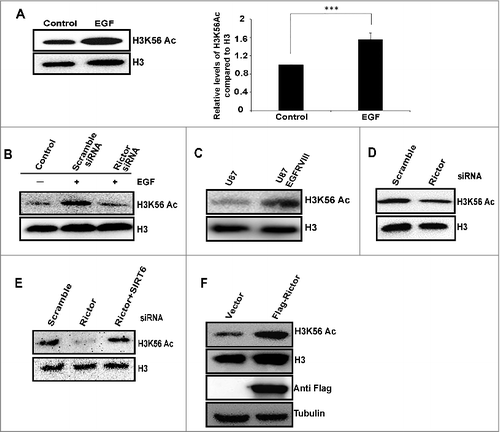
mTORC2 signaling promotes H3K56 acetylation in glioma
It has been reported that mTORC2 complex is activated by growth factors such as EGF and IGF signaling [Citation48,49]. In order to study the effect of growth factors on mTORC2 mediated regulation of H3K56Ac, we have treated HeLa cells with EGF (100 ng/10min) and observed increased H3K56Ac (). Further, rictor knockdown abrogated EGF induced H3K56Ac (), indicating that mTORC2 mediated regulation of H3K56Ac in EGF signaling pathway. Several studies reported critical functions of mTORC2 in regulating tumor growth, metabolic reprogramming, and targeted therapy resistance in Glioblastoma multiforme (GBM), which makes mTORC2 as a critical GBM drug target [Citation50]. Rictor is overexpressed and activity of mTORC2 is elevated in Gliomas [Citation51]. It has been shown that activational EGFR mutation (EGFRvIII) stimulates mTORC2 activity [Citation48]. Hence, we investigated the regulation of H3K56Ac by mTORC2 signaling in U87 and U87EGFRvIII cells (constitutively active mutant EGFRvIII). Our western blot showed increased levels of H3K56Ac in U87EGFRvIII cells than U87 cells (), suggesting upregulation of H3K56Ac in glioma by EGFR activated mTORC2 signaling. Further, knockdown of rictor resulted in decreased H3K56Ac in U87EGFRvIII cells (). Co-depletion of SIRT6 and Rictor rescued H3K56Ac levels in U87EGFRvIII cells (), suggesting SIRT6 deacetylates H3K56Ac in the absence of mTORC2 in glioma. Further, we have overexpressed rictor in U87 cells which resulted in increased H3K56Ac levels (). Overall, these results indicate that EGF pathway upregulates H3K56Ac mediated by mTORC2 in glioma.
Figure 4. mTORC2 signaling upregulates H3K56Ac in glioma. (A) EGF upregulates global H3K56Ac. HeLa cells were serum starved overnight followed by treatment with EGF (100 ng/ml) for 10 min, whole cell lysates were analyzed for levels of H3K56Ac by Western blot. Quantitative bar graph demonstrates relative H3K56Ac levels. Data are represented as mean ± SD of three independent experiments. Significant P-values were obtained with Student's t-test. ***P<0.001. (B) Knockdown of rictor abrogates EGF induced H3K56Ac in HeLa cells. HeLa cells were transfected with scramble or rictor siRNA. After 48 h, cells were serum starved overnight followed by treatment with EGF (100 ng/ml) for 10 min. Cells were harvested and whole cell lysates were analyzed for H3K56Ac by Western blot. (C) H3K56Ac levels were increased in U87EGFRvIII cells. Levels of H3K56Ac was analyzed in whole cell lysates of U87 and U87 EGFRVIII cells by western blot. (D) Knockdown of rictor downregulates H3K56Ac in U87EGFRVIII cells. U87EGFRvIII cells were transfected with scramble or rictor siRNA. After 72 h of transfection, whole cell lysates were analyzed for H3K56Ac by Western blot. (E) U87EGFRVIII cells were transfected with either scramble or rictor siRNA alone or both rictor and SIRT6 siRNA. At 72 h post transfection, whole cell lysates were analyzed for levels of H3K56Ac by Western blot. (F) overexpression of rictor in U87 cells induces H3K56Ac. U87 cells were transfected with empty vector or Flag-rictor DNA constructs and H3K56Ac was analyzed in whole cell lysates by Western blot.
mTORC2 and SIRT6 regulate glycolytic gene expression in glioblastoma
SIRT6 acts as a tumor suppressor by deacetylating H3K56 on the promoters of glycolytic genes [Citation23]. Acetylation of H3 on lysine 56 at the promoters of multiple genes promotes their transcription [Citation16,23,26,29,52]. In addition, mTORC2 has been reported to regulate the expression of glycolytic genes in U87EGFRvIII cells [Citation27]. Hence, to investigate the opposing regulation of glycolytic gene expression by mTORC2 and SIRT6 in glioma, we depleted rictor and SIRT6 individually or co-depleted rictor and SIRT6 in glioblastoma cells. As observed previously [Citation27] the expression of glycolytic genes has reduced in rictor depleted U87EGFRvIII cells (). Further, depletion of both SIRT6 and rictor rescued the expression (), indicating SIRT6 suppress glycolytic gene expression in the absence of mTORC2 in glioma.
Figure 5. mTORC2 and SIRT6 regulates expression of glycolytic genes in glioma. Rictor and SIRT6 regulate glycolytic gene expression in U87EGFRVIII cells. U87EGFRvIII cells were transfected with scramble or rictor or SIRT6 or both rictor and SIRT6 siRNA. At 48 h post transfection, cells were harvested. RNA was isolated by using Trizol method and total RNA was used for cDNA synthesis using Superscript III reverse transcriptase. mRNA levels of PDK1, LDHB, GLUT1 and LDHA genes were analyzed by RT-PCR. Data represents mean ± SEM from three independent experiments and was analyzed using a two-tailed unpaired Student's t-test. **P ≤ 0.01; *P ≤ 0.05.
mTORC2 regulates glycolytic gene expression in glioblastoma by negatively regulating SIRT6 recruitment at the promoters
Next, we investigated whether mTORC2 regulate glycolytic gene expression by regulating levels of H3K56Ac at their promoters. We performed Chromatin immunoprecipitation (ChIP) with H3K56Ac antibody followed by real-time PCR to determine the levels of H3K56Ac at the promoters of PDK1, GLUT1, LDHA and LDHB genes in U87EGFRVIII cells transfected with rictor siRNA alone or both rictor and SIRT6 siRNA. Our results revealed that levels of H3K56Ac have drastically decreased at promoters of glycolytic genes in the rictor depleted cells (), indicating that rictor knockdown leads to decreased H3K56Ac thus leading to decreased gene expression observed in . Further knockdown of SIRT6 in rictor depleted cells lead to rescued H3K56Ac on promoters of glycolytic genes (), correlating with rescued gene expression observed in . Localization of H3K9Ac at the promoters showed a modest decrease in rictor depleted cells than compared to the drastic decrease in H3K56Ac (Supplementary Figure S6). Next, to check the recruitment of SIRT6 to the promoters of glycolytic genes, we performed ChIP with SIRT6 specific antibody in rictor knockdown U87EGFRvIII cells. Our results showed that increased recruitment of SIRT6 at the promoters of glycolytic genes () in rictor knockdown cells, which correlated with decreased H3K56Ac observed in . Overall, these results suggest that mTORC2 suppresses SIRT6 recruitment to promoters of glycolytic genes resulting in increased H3K56Ac leading to their expression in glioma.
Figure 6. mTORC2 regulates expression of glycolytic genes by modulating H3K56Ac at their promoters in glioma. (A) H3K56Ac decreases on promoters of PDK1, LDHB, GLUT1 and LDHA genes in the absence of rictor. U87EGFRvIII cells were transfected with scramble, rictor or both rictor and SIRT6 siRNA. At 48 h post transfection, cells were harvested. Chromatin immunoprecipitation (ChIP) was performed using H3K56Ac antibody and primers to the promoters of PDK1, LDHB, GLUT1 or LDHA genes. (B) Localisation of SIRT6 increases on the promoters of PDK1, LDHB, GLUT1 and LDHA genes in the absence of rictor. U87EGFRvIII cells were transfected with scramble, rictor or both rictor and SIRT6 siRNA. At 48 h post transfection, cells were harvested. Chromatin immunoprecipitation (ChIP) was performed using SIRT6 antibody and primers to the promoters of PDK1, LDHB, GLUT1 or LDHA genes.
Figure 7. Model depicting the novel function of mTORC2 signaling in promoting H3K56Ac by regulating localization of SIRT6 on the promoters of glycolytic genes resulting in their elevated expression in glioma.
Discussion
The signaling mechanisms that transmit information in response to environmental changes to regulate epigenetic modifications and elicit alterations in gene expression are largely unknown [Citation53]. The target of rapamycin pathway (TOR) is the universally conserved signaling pathway regulated by environmental stimuli such as nutrients, amino acids, and growth factors to promote cell growth and proliferation [Citation1]. In yeast, TORC1 pathway regulates histone H3 and H4 acetylation on the promoters of ribosomal protein genes, affecting their expression. [Citation54,55]. However, whether mTOR signaling regulates histone modifications has not been studied much in mammals. In yeast, H3K56 acetylation is regulated by the TORC1 pathway. Genetic depletion of TCO89 or rapamycin treatment reduced global levels of H3K56Ac [Citation26]. Strikingly, the TCO89 subunit of the TORC1 is not conserved in higher eukaryotes. In a recent study, analysis of mouse fed with rapamycin has a drastic decrease in H3K56Ac as compared to other histone acetylations in the brain [Citation56], suggesting a positive correlation between mTOR signaling and H3K56Ac. In this study, we report that mTORC2 signaling positively regulates H3K56Ac and demonstrate that TSC2 acts upstream of mTORC2 to regulate H3K56Ac. We show that SIRT6 deacetylates H3K56Ac in the absence of mTORC2 signaling. Our results show increased deacetylase activity of SIRT6 on rictor knockdown. Finally, in our glioma model, we show that knockdown of mTORC2 leads to increased localization of SIRT6 resulting in decreased H3K56Ac on the promoters of glycolytic genes affecting their gene expression ().
Previous studies reported crosstalk between sirtuins and mTOR. For example, SIRT1 negatively regulates mTORC1 signaling by acting through TSC2 [Citation57]. Deacetylation of S6K1 by SIRT1 and 2 is required for phosphorylation and activation of S6K1 by mTORC1 [Citation58]. SIRT1 promoted growth and survival of neurons by negatively regulating mTORC1 signaling [Citation59]. Sirtuins 1,2,3 and 4 deacetylate rictor and inhibit mTORC2 activity [Citation60]. In this study, we identified a relationship between mTORC2 and SIRT6 pathways in regulating global H3K56Ac levels. We demonstrated that loss of H3K56Ac in mTORC2 knockdown can be rescued by knocking down SIRT6 (). SIRT6 has been reported to be phosphorylated on multiple serine residues [Citation61]. Phosphorylation of SIRT6 by AKT on serine 338 targets it to MDM2 mediated degradation [Citation62]. Whether mTORC2 directly phosphorylates SIRT6 and affects its activity has to be determined. Whether mTORC2 mediated regulation of SIRT6 has any effect on non-histone substrates of SIRT6 has to be determined in future studies.
The TSC1/TSC2 complex is a key negative regulator of the PI3K-AKT-mTOR pathway. It acts either by inhibiting mTORC1 or by positively regulating mTORC2 [Citation34,35,63]. It has been shown that loss of TSC1-TSC2 complex leads to loss of Akt phosphorylation due to defects in mTORC2 activation [Citation34]. In this study, we showed that TSC2 is required for global H3K56Ac levels (). Non-canonical mTOR independent functions of TSC complex has also been reported [Citation64]. Co-depletion of rictor and TSC2 resulted in a more pronounced decrease in H3K56Ac levels than individual knockdowns (), indicating TSC2 might function independently of mTOR signaling to regulate H3K56Ac. It would be interesting to study how TSC complex regulates SIRT6 to regulate H3K56Ac. AKT regulates the nuclear/cytoplasmic shuttling of TSC2 [Citation65]. Also, TSC2 localize to the nucleus and acts as a transcription factor by binding to the promoter of epiregulin gene and suppressing the expression [Citation66]. It will be interesting to investigate whether TSC2 localize along with mTORC2 in the nucleus to regulate the levels of H3K56 acetylation.
Growth factors regulate both mTORC1 and mTORC2. Regulation of mTORC1 by growth factors is mediated by PI3K, PDK1, AKT, and Rheb and is well understood [Citation2]. While the mechanism of growth factor mediated regulation of mTORC2 is elusive. The mTORC2-mediated phosphorylation of Akt at the hydrophobic motif (HM) site (Ser473) is activated by growth factors [Citation3–5,Citation67]. Treatment of HeLa cells with EGF has increased H3K56Ac and knockdown of rictor has abrogated EGF induced acetylation of H3K56 ( and B). Our data suggest that regulation of H3K56Ac by mTORC2 is responsive to growth factors. In recent years mTORC2 has gained significant importance due to its role in cell proliferation and survival and has been proposed as an ideal target for cancer therapy [Citation68–70]. Rictor amplification and activation has been observed in multiple cancers, resulting in increased phosphorylation of Akt at serine 473 [Citation51,67,71,72]. In a recent study, alone rictor gene amplification was shown to lead lung adenocarcinoma in 18-year-old non-smoker patient [Citation73]. In this report, overexpression of rictor in HeLa and U87 cells has induced H3K56Ac. Although it has been thought that effects of mTORC2 have been mediated majorly through Akt phosphorylation, but recent studies point to Akt independent functions of mTORC2 [Citation27,74]. These studies specify the importance of studying the functions and finding more downstream targets of mTORC2 and its specific inhibitors in cancer. In this study, we found that mTORC2 regulates H3K56Ac independent of AKT signaling.
Alteration in global levels of H4 at lysine 16 acetylation has been linked to a cancer phenotype in a variety of cancers [Citation75]. Also, acetylation of many other histone residues such as H3K9, H3K18, H4K12 and H3K27 has been altered and predict differential prognosis in cancers [Citation76]. Das et al. reported that H3K56Ac increases with grade in the gliomas [Citation17], suggesting a positive correlation between tumor grade and H3K56Ac in gliomas. However, the regulation and function of this modification in tumors is unknown. In a recent study SIRT6 overexpression in glioma cell lines induced apoptosis, thus acting as a tumor suppressor [Citation77]. In this study, by using glioma model of U87EGFRvIII, we demonstrated that increased H3K56Ac in glioma is regulated by mTORC2. Our real-time PCR results indicate that rictor depletion by siRNA reduces the expression of glycolytic genes (GLUT1, PDK1, LDHA and LDHB), while co-depletion of rictor and SIRT6 rescues the gene expression (). Further ChIP-RT PCR revealed that H3K56Ac is localized at the promoters of glycolytic genes and rictor knockdown significantly decreased H3K56Ac at the promoters and co-depletion of SIRT6 and rictor rescued acetylation levels correlating with gene expression (). In S. cerevisiae regulation of rDNA copy number is regulated by TOR complex and rapamycin induces localization of sir2 on to the rDNA locus [Citation25]. We found increased SIRT6 localization on promoters of glycolytic genes in rictor knockdown (), thus leading to decreased H3K56Ac and gene expression. SIRT6 also deacetylates H3K9Ac. However, on knockdown of rictor we observed a modest decrease in H3K9Ac at promoters compared to H3K56Ac. This might be due to the involvement of SIRT1 and SIRT2 in deacetylation of H3K56Ac, as these proteins also deacetylate H3K56Ac.
In this study, we also showed conservation of mTORC2 mediated regulation of H3K56Ac in different cell lines (). As H3K56Ac is localized on promoters of multiple genes [Citation14], it would be interesting to find whether mTORC2 regulates other signaling pathway genes by modulating H3K56Ac. Regardless of the specific mechanisms involved, mTORC2 mediated regulation of H3K56Ac likely impacts many aspects of genome regulation since this histone modification functions in transcription, DNA damage and genome stability. Downregulation of H3K56 acetylation can be used as a biomarker for mTORC2 inhibition in future studies and therapies targeting mTORC2 in glioma.
Materials and methods
Cell lines
U87 and U87EGFRvIII cell lines were a kind gift from Prof. Frank Furnari, University of California-San Diego School of Medicine, La Jolla CA. HeLa, PC3, U2OS, NIH3T3, U87, and U87EGFRvIII cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin and streptomycin in a humidified 5% CO2 incubator at 37°C.
Antibodies and reagents
Antibodies against H3K56Ac, histone H3, histone H4, SIRT6, and Raptor were obtained from Abcam (Cat.no. ab76307, ab1791, ab16483, ab88494, ab40768). H3K9Ac, H4K16Ac and mTOR antibody was purchased from Millipore (Cat.no. #07-352, #07-329, #04-385). Antibodies against Tuberin, Hamartin, Rictor, Tubulin, Akt s473, Akt, phosphoS6 (ser240/244), and S6 were obtained from Cell Signaling Technology (Cat.no. #3612, #4906, #9476, #2125, #4060, #9272, #2215, #2217). Rapamycin was purchased from Selleckchem. Wortmannin was purchased from Sigma.
Gene silencing and plasmid transfection
Small interfering RNA (siRNA) against mTOR, SIRT6, Tuberin, Hamartin, Raptor, and Rictor were obtained from Ambion (Invitrogen). All transfections were performed using Lipofectamine™ 2000 transfection reagent (Invitrogen) according to manufacturer instructions. In brief, 1 × 105 HeLa or U87EGFRvIII cells were seeded in 12 well plates and grown overnight in medium without antibiotics. Lipofectamine 2000 reagent was diluted in DMEM, siRNA was added to it and incubated at room temperature for 20 min. The Lipofectamine and siRNA complex was then added to cells and incubated for 48 to 72 h before harvesting. Transfection of TSC2, TSC2N1643S, Rictor and SIRT6 plasmids was performed with PEI transfection reagent (Polysciences) according to manufacturer instructions. Briefly, one day before transfection, HeLa cells or U87 cells (4 × 106 cells) were seeded in 10cm dishes. In a sterile tube plasmid DNA was diluted in serum-free DMEM. PEI reagent (1µg/µL) was added to diluted DNA in the ratio of 3:1 (PEI (ug): total DNA (ug)) and incubated at room temperature for 15 min. DNA/PEI mixture was added to cells and incubated for 24 h before harvesting.
Cell lysis and immunoblotting
All cell lines were lysed using NETN buffer (20mM Tris-HCl, pH8, 100 mM NaCl, 1mM EDTA, 0.5% (v/v) Nonidet P-40) containing protease and phosphatase inhibitors. Total protein was extracted by high centrifugation of the lysate and estimated by Bradford method. The whole cell lysates were boiled for 5 min in 4X SDS loading dye and resolved by SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting to detect specific proteins. The intensity of protein bands was quantified using ImageJ software (NIH, USA).
Immunofluorescence microscopy
Immunofluorescence was performed as described previously by Vempati et al. [Citation18]. Briefly, HeLa cells were grown on coverslips to 70% confluency and treated with rapamycin for 24 h followed by PBS washes. Cells were fixed with ice-cold methanol for 10 min, washed with PBS thrice and further cells were blocked for 1 h in blocking solution (3% BSA in PBS). Cells were then incubated with anti-H3K56ac (Abcam, Cat.no. ab76307) antibody diluted at 1:200 in blocking solution for 1 h at room temperature. Cells were washed thrice with PBS and incubated with Alexa Flour 488-conjugated goat anti-rabbit or Alexa Flour 647-conjugated goat anti-rabbit secondary antibodies (Thermo fisher) for 1 h and washed with PBS thrice. The cells were stained with DAPI for 5 min and washed thrice with PBS. The coverslips were mounted on slides using the mounting medium (Sigma). Fluorescence images were captured using a confocal laser scanning microscope (Leica), and the data was analyzed using LCS software (Leica).
Co-Immunoprecipitation
To study SIRT6 and rictor interaction, Co-Immunoprecipitation was performed as described [Citation29] with some modifications. Briefly, cells were washed with with 1X PBS and collected in CHAPS buffer (25 mM HEPES, 2mM EGTA, 2.5 mM MgCl2 and 0.3% CHAPS) and incubated for 20 min on rotator at 4°C followed by centrifugation at 4°C. IPs were performed by incubating supernatants (1mg) with 5 ug of SIRT6 antibody or IgG (Abcam), together with 40 µl of protein A/G beads (Calbiochem, Cat.no. IP05) overnight at 4°C. After 3 washes with CHAPS buffer containing 150 mM NaCl, the immuno-complexes were eluted by boiling in 4X SDS loading buffer for 10 min and were resolved by SDS-PAGE and detected by Western blot.
RNA isolation and quantitative real-time polymerase chain reaction
RNA was isolated by Trizol extraction method (Life Technologies). Total RNA was used for cDNA synthesis using Superscript III reverse transcriptase (Invitrogen) and the above prepared cDNA was used for RT-qPCR using EvaGreen qPCR Mastermix (GBiosciences, Cat.no. 786–858). Each sample was run in triplicate and amplification was detected using 7500 Real-Time PCR system (Applied Biosystems). Transcripts were normalized to Actin by using -ΔΔCT method [Citation78]. Primer sequences are provided in supplementary data (Supplementary Table S1).
Chromatin immunoprecipitation
For ChIP experiments with H3K56 acetylation, we followed Abcam X-ChIP protocol (http://www.abcam.com/protocols/cross-linking-chromatin-immunoprecipitation-x-chip-protocol) with some modifications. Briefly, U87EGFRvIII cells were cross-linked with 0.75% formaldehyde for 10 min at room temperature followed by incubation with 125 mM of Glycine for 5 min. Cells were washed with PBS and sonicated in the ChIP lysis buffer containing 50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 1% Sodium Deoxycholate, 0.1% SDS and protease inhibitors using Bioruptor (Diagenode). In all, 200 µg of protein supernatant was taken for each ChIP and diluted 10 times with RIPA buffer (50 mM Tris-HCl, pH8, 150 mM NaCl, 2 mM EDTA, pH 8, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitors (added freshly each time). In all, 5 µg of H3K56Ac antibody or 5 µg of IgG antibody was added to the respective samples and binding was done overnight at 4°C. Twenty microliters of protein A/G beads (Calbiochem) was added for 3 h. The beads were washed 3 times with wash buffer (0.1% SDS, 1% triton-X-100, 2 mM EDTA, pH 8, 150 mM NaCl, 20 mM Tris-HCl, pH 8) and once with final wash buffer (0.1% SDS, 1% triton-X-100, 2 mM EDTA, 500 mM NaCl, 20 mM Tris-HCl, pH 8.0). The chromatin was eluted by adding 120 ml of elution buffer (1% SDS and 100 mM NaHCO3) and reverse cross-linked by using Proteinase K at 65°C for 4 h. The DNA was extracted by phenol: chloroform and ethanol precipitation (in presence of glycogen) followed by real-time qPCR. Primer sequences are provided in supplementary data (Supplementary Table S2).
Disclosure of potential conflicts of interest
Authors declare no conflict of interest
2017CC7702R-s02.docx
Download MS Word (2 MB)Acknowledgments
We thank Prof. Frank Furnari for generously providing U87 and U87EGFRvIII cell lines. We thank Dr. MS Reddy (CDFD) for generously providing rictor plasmid construct. We thank Dr. Arun Kumar (IISC) for generously providing TSC2 plasmid construct. We thank Dr. M.D Nellist for generously providing TSC2N1643S plasmid construct. Sincere thanks to members of Laboratory of Chromatin Biology and Epigenetics.
Additional information
Funding
Related Research Data
References
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. PMID:22500797
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. PMID:19812304
- Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. PMID:16919458
- Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. PMID:15718470
- Yang Q, Inoki K, Ikenoue T, et al. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. PMID:17043309
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. PMID:18466115
- Gaubitz C, Prouteau M, Kusmider B, et al. TORC2 Structure and Function. Trends Biochem Sci. 2016;41:532–545. doi: 10.1016/j.tibs.2016.04.001. PMID:27161823
- Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203:563–574. doi: 10.1083/jcb.201306041. PMID:24385483
- Rosner M, Hengstschlager M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet. 2008;17:2934–2948. doi: 10.1093/hmg/ddn192. PMID:18614546
- Workman JJ, Chen H, Laribee RN. Environmental signaling through the mechanistic target of rapamycin complex 1: mTORC1 goes nuclear. Cell Cycle. 2014;13:714–725. doi: 10.4161/cc.28112. PMID:24526113
- Tsang CK, Liu H, Zheng XF. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle. 2010;9:953–957. doi: 10.4161/cc.9.5.10876. PMID:20038818
- Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, et al. Raptor, a positive regulatory subunit of mTOR complex 1, is a novel phosphoprotein of the rDNA transcription machinery in nucleoli and chromosomal nucleolus organizer regions (NORs). Cell Cycle. 2011;10:3140–3152. doi: 10.4161/cc.10.18.17376. PMID:21900751
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384–6391. doi: 10.1038/sj.onc.1209883. PMID:17041624
- Xie W, Song C, Young NL, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–427. doi: 10.1016/j.molcel.2009.02.004. PMID:19250903
- Tan Y, Xue Y, Song C, et al. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 2013;110:11493–11498. doi: 10.1073/pnas.1309914110. PMID:23798425
- Kong S, Kim SJ, Sandal B, et al. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem. 2011;286:16967–16975. doi: 10.1074/jbc.M111.218206. PMID:21454709
- Das C, Lucia MS, Hansen KC, et al. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. PMID:19270680
- Vempati RK, Jayani RS, Notani D, et al. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem. 2010;285:28553–28564. doi: 10.1074/jbc.M110.149393. PMID:20587414
- Battu A, Ray A, Wani AA. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res. 2011;39:7931–7945. doi: 10.1093/nar/gkr523. PMID:21727091
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. PMID:22395773
- Michishita E, Park JY, Burneskis JM, et al. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. PMID:16079181
- Kugel S, Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci. 2014;39:72–81. doi: 10.1016/j.tibs.2013.12.002. PMID:24438746
- Kugel S, Feldman JL, Klein MA, et al. Identification of and Molecular Basis for SIRT6 Loss-of-Function Point Mutations in Cancer. Cell Rep. 2015;13:479–488. doi: 10.1016/j.celrep.2015.09.022. PMID:26456828
- Cai J, Zuo Y, Wang T, et al. A crucial role of SUMOylation in modulating Sirt6 deacetylation of H3 at lysine 56 and its tumor suppressive activity. Oncogene. 2016;35:4949–4456. doi: 10.1038/onc.2016.24.
- Ha CW, Huh WK. Rapamycin increases rDNA stability by enhancing association of Sir2 with rDNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2011;39:1336–1350. doi: 10.1093/nar/gkq895. PMID:20947565
- Chen H, Fan M, Pfeffer LM, et al. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012;40:6534–6546. doi: 10.1093/nar/gks345. PMID:22553361
- Masui K, Tanaka K, Akhavan D, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. PMID:24140020
- Duvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. PMID:20670887
- Sebastian C, Zwaans BM, Silberman DM, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. PMID:23217706
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33 Suppl:245–254. doi: 10.1038/ng1089. PMID:12610534
- Workman JJ, Chen H, Laribee RN. Saccharomyces cerevisiae TORC1 Controls Histone Acetylation by Signaling Through the Sit4/PP6 Phosphatase to Regulate Sirtuin Deacetylase Nuclear Accumulation. Genetics. 2016;203:1733–1746. doi: 10.1534/genetics.116.188458. PMID:27343235
- Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. PMID:16603397
- Lee JV, Carrer A, Shah S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. PMID:24998913
- Huang J, Dibble CC, Matsuzaki M, et al. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. PMID:18411301
- Huang J, Wu S, Wu CL, et al. Signaling events downstream of mammalian target of rapamycin complex 2 are attenuated in cells and tumors deficient for the tuberous sclerosis complex tumor suppressors. Cancer Res. 2009;69:6107–6114. doi: 10.1158/0008-5472.CAN-09-0975. PMID:19602587
- Liu P, Guo J, Gan W, et al. Dual phosphorylation of Sin1 at T86 and T398 negatively regulates mTORC2 complex integrity and activity. Protein Cell. 2014;5:171–177. doi: 10.1007/s13238-014-0021-8. PMID:24481632
- Xu F, Zhang Q, Zhang K, et al. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. PMID:17889663
- Maas NL, Miller KM, DeFazio LG, et al. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–119. doi: 10.1016/j.molcel.2006.06.006. PMID:16818235
- Yuan J, Pu M, Zhang Z, et al. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009;8:1747–1753. doi: 10.4161/cc.8.11.8620. PMID:19411844
- Michishita E, McCord RA, Boxer LD, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009;8:2664–2666. doi: 10.4161/cc.8.16.9367. PMID:19625767
- Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. PMID:16439206
- McCord RA, Michishita E, Hong T, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY). 2009;1:109–121. doi: 10.18632/aging.100011. PMID:20157594
- Ghosh S, Liu B, Wang Y, et al. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep. 2015;13:1396–1406. doi: 10.1016/j.celrep.2015.10.006. PMID:26549451
- Drouet J, Delteil C, Lefrancois J, et al. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. PMID:15520013
- Michishita E, McCord RA, Berber E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. PMID:18337721
- Pan PW, Feldman JL, Devries MK, et al. Structure and biochemical functions of SIRT6. J Biol Chem. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. PMID:21362626
- Grimley R, Polyakova O, Vamathevan J, et al. Over expression of wild type or a catalytically dead mutant of Sirtuin 6 does not influence NFkappaB responses. PLoS One. 2012;7:e39847. doi: 10.1371/journal.pone.0039847. PMID:22792191
- Tanaka K, Babic I, Nathanson D, et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov. 2011;1:524–538. doi: 10.1158/2159-8290.CD-11-0124. PMID:22145100
- Yin Y, Hua H, Li M, et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Research. 2016;26:46–65. doi: 10.1038/cr.2015.133. PMID:26584640
- Wu SH, Bi JF, Cloughesy T, et al. Emerging function of mTORC2 as a core regulator in glioblastoma: metabolic reprogramming and drug resistance. Cancer Biol Med. 2014;11:255–263. PMID:25610711
- Masri J, Bernath A, Martin J, et al. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. PMID:18089801
- Kugel S, Sebastian C, Fitamant J, et al. SIRT6 Suppresses Pancreatic Cancer through Control of Lin28b. Cell. 2016;165:1401–1415. doi: 10.1016/j.cell.2016.04.033. PMID:27180906
- Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–797. doi: 10.4161/epi.6.7.16209. PMID:21636976
- Rohde JR, Cardenas ME. The tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol Cell Biol. 2003;23:629–635. doi: 10.1128/MCB.23.2.629-635.2003. PMID:12509460
- Chen H, Workman JJ, Tenga A, et al. Target of rapamycin signaling regulates high mobility group protein association to chromatin, which functions to suppress necrotic cell death. Epigenetics Chromatin. 2013;6:29. doi: 10.1186/1756-8935-6-29. PMID:24044743
- Gong H, Qian H, Ertl R, et al. Histone modifications change with age, dietary restriction and rapamycin treatment in mouse brain. Oncotarget. 2015;6:15882–15890. doi: 10.18632/oncotarget.4137. PMID:26021816
- Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5:e9199. doi: 10.1371/journal.pone.0009199. PMID:20169165
- Hong S, Zhao B, Lombard DB, et al. Cross-talk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J Biol Chem. 2014;289:13132–13141. doi: 10.1074/jbc.M113.520734. PMID:24652283
- Guo W, Qian L, Zhang J, et al. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res. 2011;89:1723–1736. doi: 10.1002/jnr.22725. PMID:21826702
- Glidden EJ, Gray LG, Vemuru S, et al. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J Biol Chem. 2012;287:581–588. doi: 10.1074/jbc.M111.304337. PMID:22084251
- Miteva YV, Cristea IM. A proteomic perspective of Sirtuin 6 (SIRT6) phosphorylation and interactions and their dependence on its catalytic activity. Mol Cell Proteomics. 2014;13:168–183. doi: 10.1074/mcp.M113.032847. PMID:24163442
- Thirumurthi U, Shen J, Xia W, et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signal. 2014;7:ra71. doi: 10.1126/scisignal.2005076. PMID:25074979
- Tee AR, Fingar DC, Manning BD, et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. PMID:12271141
- Neuman NA, Henske EP. Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol Med. 2011;3:189–200. doi: 10.1002/emmm.201100131. PMID:21412983
- Rosner M, Freilinger A, Hengstschlager M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene. 2007;26:521–531. doi: 10.1038/sj.onc.1209812. PMID:16862180
- Pradhan SA, Rather MI, Tiwari A, et al. Evidence that TSC2 acts as a transcription factor and binds to and represses the promoter of Epiregulin. Nucleic Acids Res. 2014;42:6243–6255. doi: 10.1093/nar/gku278. PMID:24748662
- Zhang F, Zhang X, Li M, et al. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010;70:9360–9370. doi: 10.1158/0008-5472.CAN-10-0207. PMID:20978191
- Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29:3733–3744. doi: 10.1038/onc.2010.139. PMID:20418915
- Guertin DA, Stevens DM, Saitoh M, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. PMID:19185849
- Hietakangas V, Cohen SM. TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer. 2008;8:282. doi: 10.1186/1471-2407-8-282. PMID:18831768
- Morrison Joly M, Hicks DJ, Jones B, et al. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer Res. 2016. doi: 10.1158/0008-5472.CAN-15-3393. PMID:27013195
- Gulhati P, Cai Q, Li J, et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin Cancer Res. 2009;15:7207–7216. doi: 10.1158/1078-0432.CCR-09-1249. PMID:19934294
- Cheng H, Zou Y, Ross JS, et al. RICTOR Amplification Defines a Novel Subset of Patients with Lung Cancer Who May Benefit from Treatment with mTORC1/2 Inhibitors. Cancer Discov. 2015;5:1262–1270. doi: 10.1158/2159-8290.CD-14-0971. PMID:26370156
- Yuan M, Pino E, Wu L, et al. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem. 2012;287:29579–29588. doi: 10.1074/jbc.M112.386854. PMID:22773877
- Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. PMID:15765097
- Khan SA, Reddy D, Gupta S. Global histone post-translational modifications and cancer: Biomarkers for diagnosis, prognosis and treatment? World J Biol Chem. 2015;6:333–345. doi: 10.4331/wjbc.v6.i4.333. PMID:26629316
- Feng J, Yan PF, Zhao HY, et al. SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress and suppression of JAK2/STAT3 signaling pathway activation. Oncol Rep. 2016;35:1395–402. doi: 10.3892/or.2015.4477. PMID:26648570
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. PMID:11846609