
ABSTRACT
DNA polymerase (pol) η is a specialized error-prone polymerase with at least two quite different and contrasting cellular roles: to mitigate the genetic consequences of solar UV irradiation, and promote somatic hypermutation in the variable regions of immunoglobulin genes. Misregulation and mistargeting of pol η can compromise genome integrity. We explored whether the mutational signature of pol η could be found in datasets of human somatic mutations derived from normal and cancer cells. A substantial excess of single and tandem somatic mutations within known pol η mutable motifs was noted in skin cancer as well as in many other types of human cancer, suggesting that somatic mutations in A:T bases generated by DNA polymerase η are a common feature of tumorigenesis. Another peculiarity of pol ηmutational signatures, mutations in YCG motifs, led us to speculate that error-prone DNA synthesis opposite methylated CpG dinucleotides by misregulated pol η in tumors might constitute an additional mechanism of cytosine demethylation in this hypermutable dinucleotide.
Introduction
The etiology of cancer lies in changes of genetic programming within the cell. Over the last decade, advances in sequencing technologies have potentiated the sequencing of whole genomes of both liquid and solid cancers (as well as individual tumor cells) giving birth to the new field of cancer genomics. One of the most significant discoveries has been that genomes of cancer cells differ from the genomes of normal cells in their immediate vicinity in terms of thousands of newly acquired cancer-driving and passenger mutationsCitation1–3, in perfect accordance to “mutator” theory of cancerCitation4,Citation5. Multiple mutagenic processes, instigated by hereditary defects, or driven by intrinsic and environmental mutagens, contribute to this “genetic collapse” that changes the identity of cellsCitation6–8. The spectrum of genetic changes includes point mutations and other micro-lesions, chromosomal rearrangements and copy number changes that can be characteristic of both cancer and tissue type. For example, different types of tumor differ strikingly between mouse strains with defective exonucleases of pol δ versus pol ϵCitation9 or when different members of APOBEC family are expressed, such as activation-induced deaminase AID (predominantly liquid tumors) versus APOBEC3B (breast and other solid tumors) in humansCitation10–12. The hereditary lack of mismatch repair and/or exonuclease activity of replicative DNA pols predispose to colorectal cancerCitation13–15; abnormal DNA double strand break repair leads to an increase in incidence of breast and ovarian cancerCitation16; sunlight and defective pol η cause skin cancerCitation17.
Normal somatic cells also acquire mutations induced by the abovementioned plethora of factors during an individual's life time, albeit at lower rates than in tumors. For instance, comparison of the mutational burden in skin fibroblasts from forearm and hip from the same donors, revealed that the UV-induced (primarily C:G > T:A and CC:GG > TT:AA changes) and endogenous mutation rates per year in exposed skin were more than two-fold higher than that in unexposed areasCitation18. This is in accord with previous studies of somatic mutations in sun-exposed skinCitation19,Citation20. Cyclobutane pyrimidine dimers and (6-4) photoproducts are the two major classes of lesions generated in DNA by UVB and UVC irradiation. Bypass of UV-induced photoproducts at TT tandem bases by the yeast and human translesion pol η (a member of the Y family of specialized DNA polymerases) is relatively accurate; this polymerase inserts the complementary AA nucleotides into the newly synthesized DNA in more than 99% of bypass events (measured using steady-state kinetic assays), thereby bypassing the lesion and suppressing the mutagenic effect of UV-induced DNA damageCitation21.
DNA pol η copies undamaged DNA with a lower fidelity than most DNA-directed polymerases with an average base-substitution error rate of 3.5 × 10−2 Citation22–24. Germline mutations in the gene (POLH) encoding DNA pol η result in XPV, a variant type of xeroderma pigmentosumCitation25. Analysis of somatic mutations has suggested that transcription-coupled repair systems and DNA pol η are involved in the control of generation of somatic mutations in normal skin cellsCitation18,Citation20. It has also been noted that the ‘pol η mutational signature’ (Signature 9; http://cancer.sanger.ac.uk/cosmic/signatures) occurs in chronic lymphocytic leukemia and malignant B-cell lymphoma genomesCitation6,Citation26,Citation27. “Signature 9” is characterized by a pattern of mutations that has been attributed to pol η (see the Discussion section for details) recruited for the repair of DNA damaged by AID during somatic hypermutation in immunoglobulin genesCitation28–30. As evident from MutaGene, (Materials and Methods, https://www.ncbi.nlm.nih.gov/research/mutagene/), mutational profiles of kidney carcinomas and esophageal adenocarcinoma, as well as signatures A.3 and B.7 also show the characteristics motif of pol η. The mutable motif of pol η, the short motif WA/TW (W = A or T) was delineated in the context of somatic hypermutations and in vitro systemsCitation22,Citation23. We detected this signature in follicular lymphomas, but only significant in 5’UTR regionsCitation27,Citation36. A recent study suggested that pol η may cause somatic mutations in lymphoid cellsCitation31; most of the characteristic clustered mutations were found in promoters, as with AID-initiated somatic hypermutation. In solid tumors, however, somatic mutations are likely to be associated with the other factors, including exogenous exposures, UV radiation or alcohol consumptionCitation31.
In this paper, we have studied the possible involvement of pol η in the generation of somatic mutations in skin cancer, other cancers and in normal cells. A highly significant correlation between pol η mutable motifs and somatic mutations in skin cancer cells was found. However, this correlation was not observed in normal skin samples. In addition to this, we also found traces of pol η mutagenesis in various other cancers. Taken together with the results of expression analysis, our study suggests the widespread participation of pol η in mutagenesis in cancer cells.
Results
Analysis of single and tandem somatic mutations found in normal skin samples
The starting point of our study was an analysis of single and tandem mutations in normal skin samples because of the known role of various DNA pols in the generation of somatic mutations in vigorously proliferating and exposed to environmental insults normal skin cellsCitation9,Citation18,Citation20. The majority of tandem double mutations are likely to be caused by the bypass of UV photoproducts formed between two pyrimidine residues, which is expected to be a significant feature of the mutational signature of pol ηCitation32–34. The dinucleotide mutabilities of CC, CT, TC and TT are actually strikingly different (). TT dinucleotides have the lowest frequency of double and single mutations, consistent with the suggested antimutagenic property (see the Introduction section) of pol η while bypassing TT dimers. CC dinucleotides are extremely susceptible to changes (mostly transitions) and yielded the largest bases of tandem double mutations ().
Figure 1. Frequency of tandem double (blue) and single mutations (red) in various dinucleotides. The Fnorm is a normalized frequency of double or single mutations (the number of mutations in dinucleotides XX multiplied by 1000 and divided by the number of dinucleotides XX in the DNA neighborhood).
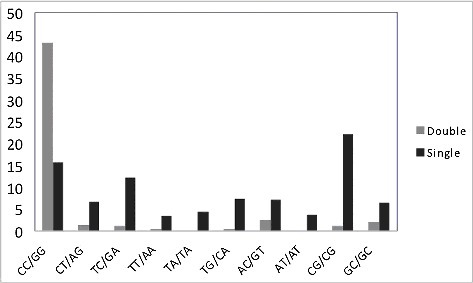
Single mutations demonstrated a different propensity: the most frequently mutated are CG dinucleotides (). It is well known that the motif YCG/CGR is hypermutated in human normal and cancer skin cellsCitation18,Citation35. CC dinucleotides were also found to be highly mutable although the frequency of mutation was lower than for CG dinucleotides (). The third highest ranked mutable dinucleotide was TC/GA. If we assume that pol η is responsible for the inaccurate bypass of dimers in CC, TC and CT dinucleotides, one would expect there to be an excess of single mutations in TC and CT dinucleotides (T is processed correctly and mutations arise while synthesizing past C nucleotides). We analyzed the excess of single mutations in TC/GA and CT/AG (positions of studied mutations are underlined). Examination of the DNA sequence context of mutations in these motifs showed that there was indeed a significant excess of substitutions (). The analysis was performed as described previouslyCitation36. In brief, we calculated the excess of mutations in specific motifs using the ratio Fm/Fs, where Fm is the fraction of mutations observed in the particular motif, and Fs is the frequency of the motif in the respective DNA neighborhood (defined as a 120 bp DNA sequence window). A 1.2-fold excess of mutations (defined as described in Materials and Methods) in TC/GA and CT/AG dinucleotides was detected (). By contrast, there was no association between mutations and the WA/TW motif, associated with predominant errors of pol η when copying undamaged DNACitation23,Citation29, indicating that pol η is unlikely to be involved in mutagenesis at undamaged DNA sites in normal skin cells ().
Table 1. Association between DNA polymerase η mutable motifs (WA/TW, TC/GA and CT/AG)Table Footnote# and the DNA sequence context of somatic mutations in normal and cancer skin cells.
Analysis of somatic mutations in skin cancer samples
Analysis of skin cancer cells strongly suggested that somatic mutations overlap with mutable motifs expected as a consequence of the error-prone bypass of photoproducts (TC/GA and CT/AG motifs) and the synthesis of undamaged DNA (WA/TW motifs) (). We also performed an analysis of two skin cancer subtypes with the highest representation in the COSMIC data set (see Materials and Methods), skin cutaneous melanoma and skin adenocarcinoma. A substantial (and significant) excess of somatic mutations for the both mutable motifs was found for skin cutaneous melanoma () where the frequency of UV photoproducts is expected to be high. However, no such excess was found for skin adenocarcinoma (), consistent with the fact that adenocarcinoma initiates in the glandular cells that are located deep inside or even under skin tissues, where no elevated frequency of UV photoproducts and mutations caused by DNA pol η in pyrimidine dinucleotides is to be expected.
Analysis of somatic mutations in cancers other than skin
Previously, we found a signature of pol η (WA/TW) in follicular lymphoma which was significant only in 5’UTR regions (P-value = 0.01)Citation36. Thus, it was suggested that a somatic mutational process operates in these regions in the “standard immunoglobulin mode” (significant correlation of mutation context with WRCH/DGYW and WA/TW mutable motifs, R = G or A, Y = C or T, D = A or T or G). The 5’UTR regions are known to be preferentially targeted by deaminases in actively transcribed genesCitation37,Citation38. This is consistent with earlier studies that suggested that pol η may be mutagenic in chronic lymphocytic leukemia and malignant B-cell lymphoma genomesCitation39. However, a more careful analysis of somatic mutations associated with pol η in follicular lymphoma suggests that this process is associated with translocations of the BCL2 gene with immunoglobulin genes, a characteristic feature of follicular lymphomaCitation40. Specifically, a detailed analysis of pol η mutability suggested that a substantial proportion (24%) of mutated 5’UTR WA/TW motifs occurred within the BCL2 gene (19 out of 28 mutations at A:T bases). After we removed mutations that were identified within the BCL2 5’UTR region (near the translocation breakpoint), the correlation became insignificant (P-value = 0.11, 60 mutations in WA/TW motifs out of 116 mutations at A:T bases)Citation27. This is one example of how one mutation hotspot (in this case resulting from a translocation) is able to skew the results of the whole exome analysis, yielding misleading results.
The discrepancies in results before and after elimination of somatic mutations associated with translocation events prompted us to analyze the pol η mutable motifs in different (sub)types of cancer. We did not find any significant excess of somatic mutations in WA/TW motifs in all types of blood cancer merged together (). However, we did find such an excess in chronic lymphocytic leukemia (CLL) and GCB lymphomas (subtypes of blood cancer) (), whereas no significant excess was found for acute myeloid leukemia (AML) (). This suggests that pol η may be mutagenic only in some types of blood cancer, consistent with the results of previous studiesCitation39.
Table 2. Preferential mutability of DNA polymerase η mutable motifs (WA/TW) in various cancers (single mutations from Whole Genomes and Whole Exomes, the Sanger COSMIC Whole Genome Project).
We found a significant excess of somatic mutations in WA/TW motifs in 11 out of 14 solid tumors from various tissue types (). Frequent tandem mutations are known to be an intrinsic property of DNA pol η when copying undamaged DNA and they have the same context specificity as single mutationsCitation23. Although tandem mutations occur much less frequently, we nevertheless found a significant excess of tandem mutations in the WA/TW context in 3 out of 8 cancer types (). A significant excess of tandem mutations in lung cancer () appears to contradict the absence of any association between single somatic mutations and the WA/TW context (). This may result from greater sensitivity of the tandem mutation analysis or from differential representation of lung cancer subtypes in datasets of single and tandem mutations. To test the latter possibility, we performed an analysis of single mutations in two non-small cell lung cancer subtypes with the highest representation in the COSMIC data set, squamous cell carcinoma and adenocarcinoma. No significant association between mutations and the WA/TW context was found in lung adenocarcinoma, whereas a significant excess (1.3, P = 0.0005) of somatic mutations in the WA/TW context was found for lung squamous cell carcinoma suggesting that DNA pol η may be involved in mutagenesis in some lung cancer subtypes but not others. It should be noted that many (although not all) lung cancers are associated with cigarette smoking and exposure to a wide variety of exogenous mutagens, any of which could influence the observed mutational spectrumCitation6–8,Citation41.
Table 3. Analysis of tandem somatic mutations in DNA polymerase η mutable motifs (WA/TW) in various cancers (Whole Genomes and Whole Exomes, the Sanger COSMIC Whole Genome Project).
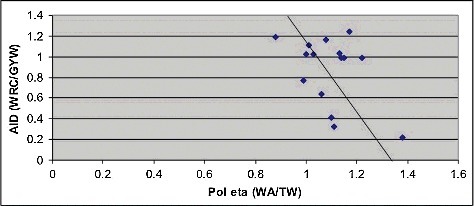
Previously, we studied the role of AID in various cancer types and found the AID mutational signature to be prevalent in many types of human cancer, suggesting that AID-mediated, CpG methylation-dependent mutagenesis is a common feature of tumorigenesisCitation36. AID and DNA pol η are the two principal mutators involved in the somatic hypermutation of immunoglobulin genes that are coupled in the hypermutation machinery: AID is involved in the initiation of somatic hypermutation by massive cytosine deamination, whereas DNA pol η in involved in error-prone repair of DNA with the resulting lesionsCitation28–30,Citation42. We proposed to analyze the possible connection between these two enzymes in various cancer types using the excess of the number of mutations in mutable motifs as independent variables. We found a negative correlation (R = −0.44) between these two variables () which suggested that AID and pol η are even decoupled in cancer-related mutagenesis (though the observed negative correlation is marginally significant, P = 0.044, one-tailed test).
Figure 2. Comparison of excess of somatic mutations associated with AID and pol η mutable motifs in various types of cancer. The excess of mutations in motifs was calculated using the ratio Fm/Fs, where Fm is the fraction of somatic mutations observed in the studied mutable motif (the number of mutated motifs divided by the number of mutations), and Fs is the frequency of the motif in the DNA context of somatic mutations (the number of motif positions divided by the total number of all un-mutated positions in surrounding regions). Linear correlation coefficient is –0.44 (P = 0.044, one-tail test). The regression line is shown in black.
Analysis of somatic mutations in various normal tissues and expression analysis of Pol η
As a control, we examined the context of somatic mutations in various normal tissuesCitation43 and did not find any significant excess of WA/TW mutable motifs (Supplemental Table 1). The size of these datasets is limited, but a power analysis (see Materials and Methods) suggested that the absence of any significant excess of somatic mutations in WA/TW mutable motifs in normal tissues likely reflects genuine biological properties of these samples.
We also compared the expression levels of the POLH gene (which encodes pol η) in various TCGA cohorts. Quartiles and extrema were calculated for each TCGA cohort selected in the study (Supplementary ). The observed high variability in POLH gene expression suggests that the gene is highly expressed only in a subset of TCGA tumor cohorts (Supplementary ) which is consistent with previous studiesCitation44. Specifically, POLH seems to be highly expressed in skin cutaneous melanoma (SKCM), consistent with a substantial and significant excess of pol η mutational signatures in this cancer type (). Previous analysis of an additional TCGA cohort with increased POLH expression, namely lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), suggested the possible involvement of pol η because of the presence of its characteristic mutation signature (Signature 9, Supplementary ,Citation39). Notably, subsets of colorectal and uterine cancer (COAD, UCEC), which have been previously reported to have no association with polymerase pol η activity, exhibit reduced POLH gene expressionCitation45,Citation46.
Discussion
A study of mutational signatures left by mutagenic enzymes, and, specifically, by pol η, can be augmented by investigating the expression profiles of the genes encoding for the enzymes in questionCitation10,Citation47. The TCGA atlas represents a comprehensive resource for the investigation of gene expression in the context of mutation datasets obtained from cohorts characterized by differing rates of somatic mutation. Observed heterogeneity in POLH gene expression within a comprehensive list of TCGA cohorts is consistent with previous reports suggesting that POLH activity is tissue- and tumor-specificCitation45. Importantly, an elevated level of POLH expression was observed in tumor cohorts where pol η-specific mutation signatures were detected. Conversely, a reduced level of POLH expression was observed in tumor cohorts where no pol η-specific mutation signatures were detected.
The excess of pol η mutable motifs in chronic lymphocytic leukemia and GCB lymphomas that we detected in our work is consistent with the studies of Alexandrov et al.Citation39 where the pol η signature, “Signature 9”, was detected in chronic lymphocytic leukemia and malignant B-cell lymphoma genomes. This is a promising result bearing in mind that the mutable motif of pol η (WA/TW) is rather short and hence less informative as compared to the AID/APOBEC mutable motifsCitation27,Citation29. In general, “Signature 9” is characterized by a pattern of mutations that has been widely attributed to pol η, although a higher frequency of T:A > G:C transversions compared to T:A > C:G transitions, has not been observed in studies of pol η either in vitro or in vivoCitation27,Citation29. The decomposition into signatures is a very useful tool for interpreting mutagenic processes, this approach has certain limitationsCitation27. One of them is the heuristic nature of the associations between mutational signatures and molecular mechanisms of mutation. In fact, we can never be sure that a given mutational signature can be attributed solely and exclusively to one molecular mechanism – indeed, some endogenous or exogenous mutational mechanisms may have very similar or even identical signaturesCitation27,Citation29.
It should be stressed that important steps toward improving our understanding of the role of pol η in mutagenesis in skin cancer have been taken in previous studies, where the impact of transcription-coupled repairCitation20 and pol ηCitation18 in both normal and cancer skin cells were postulated. It should be noted that the strand-specificity (a signature of transcription-coupled repair) of mutations induced by pol η is well known in the context of the somatic hypermutation of immunoglobulin genesCitation18,Citation20,Citation48,Citation49. Thus, all these studies point to pol η being an important mutagenic factor in normal skin and cancer cells. The recent study extended a range of potential mutagenic activity of pol η to solid tumors where somatic mutations produced by pol η are likely to be associated with the other factors, including exogenous exposures, UV radiation or alcohol consumptionCitation31.
We detected overlaps between pol η signatures with somatic mutations in various cancers. It is possible that the perturbed cell metabolism leads to the aberrant regulation of pol η, for example, that what one expects in the completely disorganized environment of cell extracts (where the pol η mutagenesis had beed inferred)Citation50, or in in vitro systemsCitation22, while normal cells are well protected from its actionCitation51. The error-prone action of misregulated pol η is expected to cause a substantial load of somatic mutations, which may be beneficial for cancer initiation and/or progression, for example, when TP53 is mutatedCitation27,Citation52. Another potential function of pol η in cancer cells could be the error-free or error-prone bypass of various DNA lesions. It was suggested in a recent paperCitation53 that NPM1 (nucleophosmin) regulates translesion DNA synthesis (TLS) via an interaction with the catalytic core of pol η. NPM1 deficiency causes a TLS defect due to the proteasomal degradation of pol η. The prevalent NPM1 mutation (c+) leading in one-third of AML patients to NPM1 mislocalization results in a loss of pol η, which may explain why no significant excess of mutation in pol η motifs was found for acute myeloid leukemia (). These results hint at the complexity of regulation of pol η in cancer cells and provide an explanation of why pol η mutational signatures are found only in some cancer types/subtypes.
It was suggested that, in both normal and cancer skin cells, a significantly increased frequency of UVB-induced transition mutations at YCG motifs could be explained by the participation of pol ηCitation35. Taking into account the high frequency of mutations in TC dinucleotides, it is tempting to speculate that mutagenesis of YCG motifs is caused by the error-prone synthesis by pol η on methylated cytosine in TCG or CCG sequences (with or without neighboring photoproducts) (). Thus, the error-prone synthesis in YCG motif might be an additional mechanism of demethylation, by pol η misincorporating A instead of G in various types of cancer cell. The evidence for that comes from the observed negative correlation between the excess of somatic mutations associated with AID and pol η mutable motifs in various types of cancer (). However, this hypothesis requires further experimental validation and would require analysis of methylated templates using in vitro pol η mutagenesis systems. The analysis of mammalian model species and cell cultures might also provide the means to test this hypothesis.
Materials and methods
Analysis of somatic mutations
DNA sequences surrounding the mutated nucleotide represent the mutation context. We compared the frequencies of known mutable motifs for somatic mutations with the frequencies of these motifs in the vicinity of the mutated nucleotides. Specifically, for each base substitution, the 120 bp sequence centered around the mutation was extracted (the DNA neighborhood). We used only the nucleotides immediately surrounding the mutations because pol η is thought to scan a limited length of DNA to mutate nucleotides in a preferred motifCitation36,Citation54. This approach does not exclude any given region of the genome in general, but rather uses the areas within each sample where mutagenesis has happened (taking into account the variability in mutation rates across the human genome), and then evaluates whether the mutagenesis in this sample was enriched for DNA pol η motifsCitation36,Citation54. This approach was thoroughly tested and its high accuracy demonstratedCitation36. The frequencies of mutable motifs in the locations of somatic mutations was compared to the frequencies of the same motifs in the DNA neighborhood () using Fisher's exact test (2 × 2 table, 2-tail test) as previously describedCitation36,Citation54 (for details see ).
Figure 3. Statistical analysis of mutable motifs in sites of somatic mutations and surrounding regions. The excess of mutations in motifs was calculated using the ratio Fm/Fs, where Fm is the fraction of somatic mutations observed in the given mutable motif (the number of mutated motifs divided by the number of mutations), and Fs is the frequency of the motif in the DNA neighborhood of somatic mutations (the number of motif positions divided by the total number of all un-mutated positions in the 120 bp window).

The exome sequencing data of somatic mutations in normal skin cells were obtained fromCitation20. Somatic mutation data from the ICGC and TCGA cancer genome projects were extracted from the Sanger COSMIC Whole Genome Project v75 http://cancer-beta.sanger.ac.uk/cosmic. The tissues and cancer types for studies of mutation signatures were defined according to primary tumor site and cancer genome sequencing projectsCitation55. Somatic mutations in various normal tissues were from Yadav et al.Citation43.
Power analysis of mutations in normal tissues
We compared the magnitude of the difference between the fraction of mutations observed in the mutable motif and the fraction of motifs in the surrounding region (effect size) for somatic mutations in normal tissues. For the purpose of this comparison (power analysis), we used a sampling procedure that was repeated 1,000 times. Each sample of all available somatic mutations from cancer cells (where a significant excess of somatic mutations in WA/TW motifs was observed, the last row in the ) had a size equal to that for normal tissues (552 somatic mutations, the last row in the Supplementary ). Analysis of the difference between the fractions showed that the difference for normal mutations was smaller for 99.1% of cancer samples. Thus, the observed effect size (Supplementary ) is likely to reflect the biological properties of these samples and is unlikely to result from the small sample size, at least for somatic mutations from normal tissues.
Expression analysis of the POLH gene
For the POLH gene expression analysis, the normalized version of the RSEM (Broad Institute TCGA Genome Data Analysis Center, 2016, Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run. Broad Institute of MIT and Harvard Dataset http://doi.org/10.7908/C11G0KM9) was used to analyze the TCGA RNA-Seq datasets from the Broad Genome Data Analysis Center. For each TCGA cohort (Supplementary ), the low and upper bounds, median, outliers, and first and third quartiles were retrieved via the FireBrowse RESTful API (http://firebrowse.org/api-docs/) for the tumor and the corresponding normal (when available) tissue samples.
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
1404208-suppl.rar
Download (121.4 KB)Additional information
Funding
Related Research Data
References
- Watson IR, Takahashi K, Futreal PA, et al. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14:703–718. doi:10.1038/nrg3539. PMID:24022702
- Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi:10.1038/nature14169. PMID:25719666
- Wood LD, Hruban RH. Genomic landscapes of pancreatic neoplasia. J Pathol Transl Med. 2015;49:13–22. doi:10.4132/jptm.2014.12.26. PMID:25812653
- Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. PMID:4136142
- Loeb LA. Human cancers express a mutator phenotype: hypothesis, origin, and consequences. Cancer Res. 2016;76:2057–2059. doi:10.1158/0008-5472.CAN-16-0794. PMID:27197248
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi:10.1038/nature12477. PMID:23945592
- Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. doi:10.1038/nrc3816. PMID:25568919
- Pfeifer GP. How the environment shapes cancer genomes. Curr Opin Oncol. 2015;27:71–77. doi:10.1097/CCO.0000000000000152. PMID:25402978
- Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106:17101–17104. doi:10.1073/pnas.0907147106. PMID:19805137
- Burns MB, Lackey L, Carpenter MA, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi:10.1038/nature11881. PMID:23389445
- Heintel D, Kroemer E, Kienle D, et al. High expression of activation-induced cytidine deaminase (AID) mRNA is associated with unmutated IGVH gene status and unfavourable cytogenetic aberrations in patients with chronic lymphocytic leukaemia. Leukemia. 2004;18:756–762. doi:10.1038/sj.leu.2403294. PMID:14961036
- Qian J, Wang Q, Dose M, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–1537. doi:10.1016/j.cell.2014.11.013. PMID:25483777
- Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5:382–395. doi:10.1016/0959-437X(95)80055-7.
- Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24:107–113. doi:10.1016/j.gde.2013.12.005.
- Barbari SR, Shcherbakova PV. Replicative DNA polymerase defects in human cancers: consequences, mechanisms, and implications for therapy. DNA Repair (Amst). 2017;56:16–25. doi:10.1016/j.dnarep.2017.06.003. PMID:28687338
- Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res: BCR. 2000;2:324–230. doi:10.1186/bcr76. PMID:11250724
- Masutani C, Kusumoto R, Yamada A, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi:10.1038/21447. PMID:10385124
- Saini N, Roberts SA, Klimczak LJ, et al. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet. 2016;12:e1006385. doi:10.1371/journal.pgen.1006385. PMID:27788131
- Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi:10.1073/pnas.0912629107. PMID:20080596
- Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Sci (New York, NY). 2015;348:880–886. doi:10.1126/science.aaa6806.
- Washington MT, Johnson RE, Prakash L, et al. Accuracy of lesion bypass by yeast and human DNA polymerase eta. Proc Natl Acad Sci U S A. 2001;98:8355–8360. doi:10.1073/pnas.121007298. PMID:11459975
- Matsuda T, Bebenek K, Masutani C, et al. Low fidelity DNA synthesis by human DNA polymerase h. Nature. 2000;404:1011–1013. doi:10.1038/35010014. PMID:10801132
- Matsuda T, Bebenek K, Masutani C, et al. Error rate and specificity of human and murine DNA polymerase eta. J Mol Biol. 2001;312:335–346. doi:10.1006/jmbi.2001.4937. PMID:11554790
- Pavlov YI, Shcherbakova PV, Rogozin IB. Roles of DNA polymerases in replication, repair, and recombination in eukaryotes. Int Rev Cytol. 2006;255:41–132. doi:10.1016/S0074-7696(06)55002-8. PMID:17178465
- Johnson RE, Kondratick CM, Prakash S, et al. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science (New York, NY). 1999;285:263–265. doi:10.1126/science.285.5425.263.
- Petljak M, Alexandrov LB. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis. 2016;37:531–540. doi:10.1093/carcin/bgw055. PMID:27207657
- Rogozin IB, Pavlov YI, Goncearenco A, et al. Mutational signatures and mutable motifs in cancer genomes. Brief Bioinform. 2017. doi:10.1093/bib/bbx049. PMID:28498882
- Zanotti KJ, Gearhart PJ. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair (Amst). 2016;38:110–116. doi:10.1016/j.dnarep.2015.11.011. PMID:26719140
- Rogozin IB, Pavlov YI, Bebenek K, et al. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi:10.1038/88732. PMID:11376340
- Zeng X, Winter DB, Kasmer C, et al. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–541. doi:10.1038/88740. PMID:11376341
- Supek F, Lehner B. Clustered mutation signatures reveal that error-prone DNA repair targets mutations to active genes. Cell. 2017;170:534.e23–47.e23. doi:10.1016/j.cell.2017.07.003.
- McCulloch SD, Kokoska RJ, Masutani C, et al. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi:10.1038/nature02352. PMID:14999287
- Saribasak H, Maul RW, Cao Z, et al. DNA polymerase zeta generates tandem mutations in immunoglobulin variable regions. J Exp Med. 2012;209:1075–1081. doi:10.1084/jem.20112234. PMID:22615128
- Maul RW, MacCarthy T, Frank EG, et al. DNA polymerase iota functions in the generation of tandem mutations during somatic hypermutation of antibody genes. J Exp Med. 2016;213:1675–1683. doi:10.1084/jem.20151227. PMID:27455952
- Lee DH, Pfeifer GP. Deamination of 5-methylcytosines within cyclobutane pyrimidine dimers is an important component of UVB mutagenesis. J Biol Chem. 2003;278:10314–10321. doi:10.1074/jbc.M212696200. PMID:12525487
- Rogozin IB, Lada AG, Goncearenco A, et al. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci Rep. 2016;6:38133. doi:10.1038/srep38133. PMID:27924834
- Lada AG, Kliver SF, Dhar A, et al. Disruption of transcriptional coactivator Sub1 leads to genome-wide re-distribution of clustered mutations induced by APOBEC in active yeast genes. PLoS Genet. 2015;11:e1005217. doi:10.1371/journal.pgen.1005217. PMID:25941824
- Taylor BJ, Wu YL, Rada C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. Elife. 2014;3:e03553. doi:10.7554/eLife.03553. PMID:25237741
- Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24:52–60. doi:10.1016/j.gde.2013.11.014.
- Green MR, Kihira S, Liu CL, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112:E1116–E1125. doi:10.1073/pnas.1501199112. PMID:25713363
- Temiz NA, Donohue DE, Bacolla A, et al. The somatic autosomal mutation matrix in cancer genomes. Hum Genet. 2015;134:851–864. doi:10.1007/s00439-015-1566-1. PMID:26001532
- Laffleur B, Denis-Lagache N, Peron S, et al. AID-induced remodeling of immunoglobulin genes and B cell fate. Oncotarget. 2014;5:1118–1131. doi:10.18632/oncotarget.1546. PMID:24851241
- Yadav VK, DeGregori J, De S. The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res. 2016;44:2075–2084. doi:10.1093/nar/gkw086. PMID:26883632
- Makridakis NM, Reichardt JK. Translesion DNA polymerases and cancer. Front Genet. 2012;3:174. doi:10.3389/fgene.2012.00174. PMID:22973298
- CGAN. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi:10.1038/nature11252. PMID:22810696
- CGARN. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi:10.1038/nature10166. PMID:21720365
- Roberts SA, Lawrence MS, Klimczak LJ, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi:10.1038/ng.2702. PMID:23852170
- Pavlov YI, Rogozin IB, Galkin AP, et al. Correlation of somatic hypermutation specificity and A-T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci U S A. 2002;99:9954–9959. doi:10.1073/pnas.152126799. PMID:12119399
- Mayorov VI, Rogozin IB, Adkison LR, et al. DNA polymerase eta contributes to strand bias of mutations of A versus T in immunoglobulin genes. J Immunol. 2005;174:7781–7786. doi:10.4049/jimmunol.174.12.7781.
- Bebenek K, Matsuda T, Masutani C, et al. Proofreading of DNA polymerase eta-dependent replication errors. J Biol Chem. 2001;276:2317–2320. doi:10.1074/jbc.C000690200. PMID:11113111
- Pavlov YI, Nguyen D, Kunkel TA. Mutator effects of overproducing DNA polymerase eta (Rad30) and its catalytically inactive variant in yeast. Mutat Res. 2001;478:129–139. doi:10.1016/S0027-5107(01)00131-2. PMID:11406177
- Lerner LK, Francisco G, Soltys DT, et al. Predominant role of DNA polymerase eta and p53-dependent translesion synthesis in the survival of ultraviolet-irradiated human cells. Nucleic Acids Res. 2017;45:1270–1280. doi:10.1093/nar/gkw1196. PMID:28180309
- Ziv O, Zeisel A, Mirlas-Neisberg N, et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat Commun. 2014;5:5437. doi:10.1038/ncomms6437. PMID:25421715
- Chan K, Roberts SA, Klimczak LJ, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47:1067–1072. doi:10.1038/ng.3378. PMID:26258849.
- Goncearenco A, Rager SL, Li M, et al. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017;45:W514–W522. doi:10.1093/nar/gkx367. PMID:28472504.