
ABSTRACT
MLN4924 inhibits the cullin-RING ligases mediated ubiquitin-proteasome system, and has showed antitumor activities in preclinical studies, but its effects and mechanisms on pancreatic cancer (PC) remains elusive. We found that MLN4924 inhibited the proliferation and clonogenicity of PC cells, caused DNA damage, particularly double-strand breaks, and leaded to Chk1 activation and cell-cycle arrest. Chk1 inhibitor SCH 900776 alone exhibited minimal cytotoxicity, and caused no DNA damage on PC cells. But in the combination therapy, SCH 900776 enhanced the cytotoxicity and DNA damage caused by MLN4924, likely by abrogating G2/M arrest and promoting DNA re-replication. In vivo study on a xenograft PC mouse model also showed that SCH 900776 increased the efficacy of MLN4924. We also evaluated the level of NEDD8-activating enzyme (NAE), the direct target of MLN4924, and found that NAE level was elevated in PC tissues compared with normal pancreas, but was irrelevant with prognosis. Our findings provide the preclinical evidence and the rationale of the combination therapy of MLN4924 with SCH 900776 or other Chk1 inhibitors to treat PC.
Introduction
Pancreatic cancer is highly malignant and ranked as the 4th leading cause of cancer-related deaths in the US [Citation1] and 6th in China [Citation2]. Gemcitabine had long been the standard chemotherapy [Citation3], and recent clinical trials gave rise to more effective regimens such as FOLFIRINOX [Citation4] and gemcitabine plus nab-paclitaxel [Citation5]. The past decades have witnessed a surge in the development and application of targeted therapies in a wide range of malignancies [Citation6–10], but regarding to pancreatic cancer, only erlotinib showed modest efficacy combined with gemcitabine [Citation11], it is thus greatly needed to find novel targeted therapies for pancreatic cancer.
The ubiquitin-proteasome system (UPS) is the major intracellular pathway for extralysosomal protein degradation with important roles in a broad array of cellular processes, such as cell growth, division and apoptosis [Citation12]. Bortezomib (Velcade), the proteasome inhibitor, is approved for the treatment of patients with multiple myeloma [Citation13] or mantle cell lymphoma [Citation14], suggesting the UPS inhibition as an attractive antitumor approach. Proteins are targeted for degradation within the UPS by conjugation with ubiquitin via a three-step cascade mechanism, in which E3 ubiquitin ligases mediate the last step [Citation15]. The cullin-RING ligases (CRLs) are the largest family of E3 ligases [Citation16]. The activation of CRLs requires the covalent binding of NEDD8 to the cullin proteins, which function as core scaffolds in the CRLs [Citation17]. NEDD8 is a small ubiquitin-like protein that is activated for conjugation by NEDD8-activating enzyme (NAE) [Citation18], thus inhibiting NAE would in turn inhibit the CRLs-mediated UPS.
MLN4924 (TAK-924/Pevonedistat) is a first-in-class highly-selective NAE inhibitor [Citation19] that has been evaluated in several phase I and phase I/II clinical trials. By generating the inhibitory NEDD8-MLN4924 covalent adduct in situ in the presence of enzymatically active NAE, MLN4924 prevents NAE from processing NEDD8 for CRLs conjugation, resulting in CRLs inhibition and substrates accumulation [Citation20]. MLN4924 shows antitumor activities toward a variety of solid and hematologic malignancies in vitro and on mouse models [Citation19,Citation21–28], possibly by inducing DNA re-replication resulted from the accumulation of CRLs substrate Cdt1 [Citation19,Citation21,Citation27], while alternative pathways are also proposed [Citation23–26,Citation28]. As it is observed in several studies [Citation19,Citation22,Citation27,Citation29] and in our study that Chk1 was activated and cell-cycle progression was arrested after MLN4924 treatment, we wonder if combining MLN4924 with Chk1 inhibitor could achieve better antitumor efficacy, especially toward pancreatic cancer. We used SCH900776, a potent and functionally selective Chk1 inhibitor [Citation30], to test our hypothesis. Indeed, we found SCH900776 enhances the antitumor activity and the DNA damage effects of MLN4924, likely by abrogating the G2/M arrest and reinforce DNA re-replication.
Materials and methods
Cell lines and chemicals
Human pancreatic cancer cell lines Hup-T3 was purchased from Sigma Aldrich and cultured in EMEM (EBSS) + 2mM Glutamine + 1% non-essential amino acids + 1% sodium pyruvate + 10% Fetal Bovine Serum (FBS). Human pancreatic cancer cell lines PANC-1 was obtained from the American Type Culture Collection (ATCC) and cultured in DMEM + 10% FBS. All cells were cultured at 37°C with 5% CO2. MLN4924 was purchased from Millipore (951950-33-7). For in vitro studies, MLN4924 was dissolved in dimethyl sulfoxide (DMSO) and stored in −20°C. For in vivo studies, MLN4924 was dissolved in 10% 2-hydroxypropyl-β-cyclodextrin (HPBCD) and stored in at 4°C. SCH 900776 was purchased from Cayman chemical (891494-63-6). For in vitro studies, SCH 900776 was dissolved in dimethyl sulfoxide (DMSO) and stored in −20°C. For in vivo studies, SCH 900776 was dissolved in 20% HPBCD and stored in at 4°C.
Cytotoxicity and clonogenic survival assays
Hup-T3 cells were seeded into 96-well plates at 1000 cells per well and allowed to adhere for 24 hours. Cells were then treated with different chemicals at various concentrations for 72 hours. Cell viability was assessed with CellTilter 96 AQueous One Solution Cell Proliferation Assay (MTS) according to the manufacturer's instructions (Promega). Briefly, 20 ml of MTS were added to each well 3 hours later. The absorbance at 490 nm was measured with a microplate reader (Tecan Infinite M1000 PRO).
300 cells were plated in triplicate in 6-well plates. After 24 hours, different chemicals were added into the medium to reach various concentrations. Medium was changed every 2–3 days and chemicals were then added to maintain the previous concentrations. After 10 d of incubation, the colonies were fixed with ice-cold methanol and stained with 0.05% crystal violet. Colonies containing >50 cells were counted.
Antibodies and immunoblotting
DNA Damage Antibody Sampler Kit (#9947) was from Cell Signaling Technology, including rabbit antibodies against phospho-Chk2 (Thr68), phospho-Chk1 (Ser345), phospho-BRCA1 (Ser1524), phospho-Histone H2A.X (Ser139), phospho-ATM (Ser1981). Anti-phospho-Chk1 (Ser296) antibody was kindly provided by Dr. Karnitz, Mayo Clinic. Antibodies against phospho-KAP1 (Ser824), MDC1 and 53BP1 were generated in Dr. Zhenkun Lou's lab. Anti-NEDD8 antibody (ab209076) and anti-histone H3 (ab1791) were from Abcam, anti-β-actin was from Sigma (A2228). Fluorescent dye–conjugated secondary antibodies (A-11001, A-11034, 31660, 31686) were obtained from Invitrogen. Horseradish peroxidase–conjugated secondary antibodies against mouse (sc-2748) and rabbit (sc-2750) were obtained from Santa Cruz.
Cells were lysed with 0.5% NP40, 150 mM NaCl, 50 mM Tris, and 1 mM EDTA (NETN) buffer with 50 mM β-glycerophosphate, 10 mM NaF, and 1 mg/ml each of pepstatin A and aprotinin. Proteins were separated by SDS-PAGE, transferred onto PVDF membrane and probed using appropriate primary and secondary antibodies. Protein detection was performed with ECL reagents (ThermoFisher).
Immunofluorescence staining
Cells were seeded on coverslips and fixed with 3% paraformaldehyde for 10 minutes, then permeabilized in 0.5% Triton-X for 5 minutes at room temperature. Cells were blocked with 5% goat serum at room temperature for 1 hour. Then cells were incubated with primary antibody at 37°C for 20 minutes, followed by incubation with Alexa Fluor 488 or rhodamine-conjugated secondary antibodies at 37°C for 20 minutes. Cell nuclei were counterstained with DAPI. Slides were washed with PBS between incubations.
Propidium iodide staining and flow cytometric analysis
Cells were fixed with 70% ethanol at −20°C overnight, stained with propidium iodide containing RNase at 37°C for 15 minutes, then analyzed for cell-cycle profile by CyAn ADP (Beckman Coulter). Data were analyzed with ModFit LT software.
In vivo antitumor study
This study was approved by the Animal Ethics Committee of Fudan University, and all animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of Fudan University and the National Institutes of Health guide for the care and use of Laboratory animals. 1 × 106 Panc-1 cells were inoculated subcutaneously in the left flank of nude mice. When the tumor reached approximately 100 mm3 in volume, the tumor-bearing mice were randomized into the following 4 groups (8 animals/group) and treated with (1) 20% HPBCD, (2) MLN4924 (30 mg/kg, s.c.), (3) SCH 900776 (8 mg/kg, i.p.), (4) MLN4924 combined with SCH 900776 at the preceding dosage, on a five days on and two days off schedule for two weeks. Mice were weighed and tumor length and width were measured every other day. Tumor volume was estimated from the formula: length × width2/2. We use tumor growth inhibition (TGI) to evaluate the efficacy of the treatment according to the following formula: TGI (%) = [100 – (Ti – T0)/ (Ci – C0) × 100]; Ti and Ci represent the average tumor volume of the treatment group and the control group at the end of study, respectively; T0 and C0 represent the average tumor volumes at the initiation of the treatment.
Patients' specimens and follow-up
The study was approved by the Zhongshan Hospital Research Ethics Committee and informed consent was obtained from all patients. 90 cases of patients that underwent radical surgeries during 2012–2014 in Zhongshan Hospital with pathological diagnosis of pancreatic ductal adenocarcinoma were selected, and the medical histories of the patients were collected. None of the patients received preoperative anticancer treatment, and all of them received standard postoperative chemotherapy of gemcitabine. Follow-ups were conducted via phone, mail or return visits, and all patients were followed up until February 2017.
Immunohistochemical staining and expression scoring
Paraffin-embedded pancreatic cancer tissue and normal pancreas tissue blocks of the selected patients were retrieved and tissue microarrays were made in the way as decribed earlier [Citation31]. The slides were incubated overnight with the primary antibody, followed by incubation with the secondary antibody. Diaminobenzidine chromogenic substrate system was used for colorimetric visualization. The density of NAE staining was scored according to the staining intensity (negative, weak, moderate or strong) and fraction of stained cells (<25%, 25–75% or >75%). Each combination of intensity and fractions is automatically converted into a protein expression level score as follows: negative – not detected; weak <25% – not detected; weak combined with either 25 – 75% or 75% – low; moderate <25% – low; moderate combined with either 25 – 75% or 75% – medium; strong <25% – medium, strong combined with either 25 – 75% or 75% – high.
Statistical analysis
Data of individual experiments were averaged and presented as mean ± SD. Student t-test and ANOVA were used to compare quantitative variables. The expression levels of NAE was compared using Wilcoxon signed-rank test and chi-square test. Kaplan–Meier analysis was used to determine the survival and log-rank test was used to compare survival of patients.
Results
SCH 900776 enhanced the cytotoxicity of MLN4924 on pancreatic cancer cells
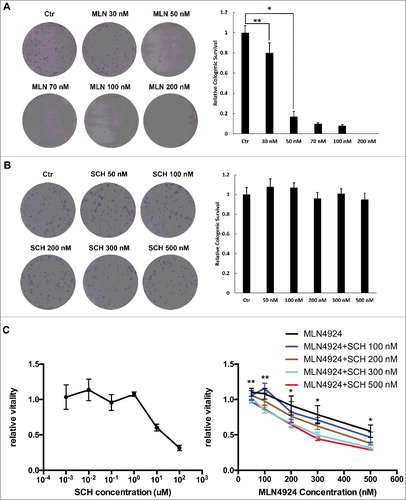
After Hup-T3 cells were treated with of MLN4924 and SCH 900776 alone or in combination, clonogenic assay and MTS assay were performed to evaluate cell survival and proliferation. As shown in A, MLN4924 at 50 nM inhibited the colony formation effectively in Hup-T3 cells, while SCH 900776 didn't affect the colony formation even at 500 nM (B). Also, MLN4924 significantly inhibited the proliferation of Hup-T3, in contrast SCH 900776 didn't suppress the proliferation until at 10 uM (C). But when in combination, at low concentrations SCH 900776 enhanced the cytotoxity of MLN4924, and the enhancement increased with the concentration of SCH 900776 (P<0.05) (C).
Figure 1. SCH 900776 enhances the cytotoxicity of MLN4924 on pancreatic cancer cells. A and B, MLN 4924 and SCH 900776 on the clonogenicity of pancreatic cancer cells. 300 cells were seeded in triplicate in 6-well plates and treated with various concentrations of MLN4924 (A) and SCH 900776 (B) respectively for 10 days, and colonies with more than 50 cells were counted. Left, representative images of the results; right, relative colony numbers compared with the control (mean ± SD). C, MLN 4924 and SCH 900776 on the proliferation of pancreatic cancer cell. Cells were seeded into 96-well plates at 1000 cells per well and were then treated with SCH 900776 (left), MLN4924 and SCH 900776 (right) at various concentrations for 72 hours. Cell viability was assessed with MTS assay. *, P<0.05; **, P>0.05; MLN, MLN4924; SCH, SCH 900776.
MLN4924 caused DNA damage on pancreatic cancer cells
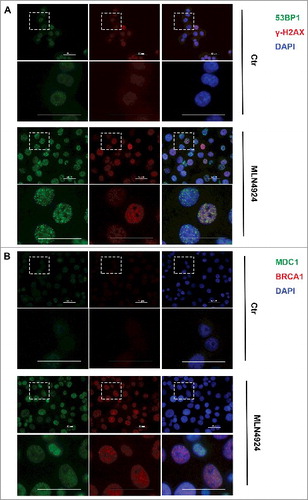
It is reported that MLN4924 treatment caused tumor cell DNA damage proved by elevated level of phosphorylated Chk1 (Ser 317) [Citation19,Citation27]. We then measured the overall levels of phosphorylated Chk1, phosphorylated ATM, and phosphorylated Chk2 as markers of DNA damage. We found that MLN4924 at 300 nM for a 4-hour exposure was sufficient to block neddylation of cullins, but the DNA damage markers were activated at a later time point (24 hours) (). To determine the DNA damage response upon MLN4924 treatment directly and more thoroughly, we measured the formation of nuclear foci of several proteins involved in the DNA damage repair pathway (γ-H2AX, MDC1, 53BP1, BRCA1). After treated with MLN4924 at 300 nM for 24 hours, foci of each protein were formed in almost all Hup-T3 cells (), suggesting that MLN4924 treatment causes DNA damage. Meanwhile, MLN4924 didn't cause the formation of foci or the overall expression change of ATM, CHK1, CHK2, cullin-1 and cyclin D1 (Supplementary Figure).
Figure 2. MLN4924 inhibits the neddylation of cullins and causes DNA damage on pancreatic cancer cells. Hup-T3 cells were treated with MLN4924 at 300 nM for indicated time periods, followed by IB with β-actin as a loading control. MLN, MLN4924.
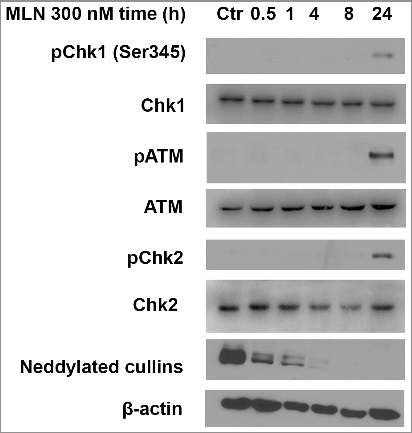
Figure 3. MLN4924 causes double-strand breaks on pancreatic cancer cells. A and B, Hup-T3 cells were treated with MLN4924 at 300 nM for 24 hours, followed by immunofluorescence staining for 53BP1 and γ-H2AX (A), or MDC1 and BRCA1 (B). Scale bar, 50 μm.
SCH 900776 enhanced the DNA damage caused by MLN4924 by abrogating G2/M arrest and promoting DNA re-replication
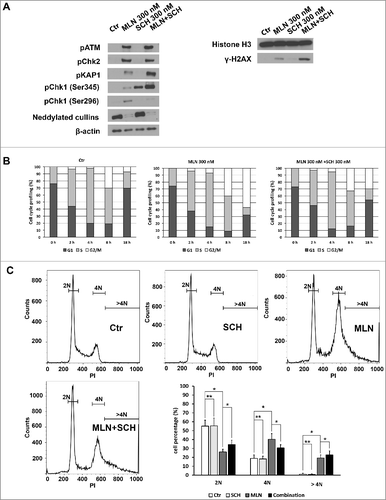
Since almost all cells were foci positive after MLN4924 treatment, it is impossible to evaluate the potential enhancing effect of SCH 900776 by immunofluorescence, so we used Western blot to test the levels of DNA damage markers. Compared with MLN4924 alone, higher levels of phosphorylated Chk1, phosphorylated ATM and γ-H2AX were observed in cells treated by MLN4924 plus SCH 900776 for 24 hours (A, suggesting more DNA damage in cells treated with MLN4924 plus SCH 900776. To investigate the underlying mechanisms of the enhancement, we assessed the cell-cycle profile of pancreatic cancer cells treated with MLN4924, SCH 900776 or the combination. First, we discovered that MLN4924 induced G2/M arrest in Hup-T3 cells, as most of the cells (58%) were still in the G2/M phase 18 hours after release from the thymidine block (b). Compared with MLN4924 alone, the addition of SCH 900776 partly abrogated the MLN4924-induced G2/M arrest, as cells in the G2/M decreased (30.6% vs. 40.3%, P<0.05) and cells in G1 increased (34.3% vs. 26.1%, P<0.05) (c). Meanwhile cells with DNA contents greater than 4N also increased (22.7% vs. 19.3%, P<0.05) (c). This is consistent with a role of Chk1 in the G2/M checkpoint.
Figure 4. SCH 900776 enhances the DNA damage caused by MLN4924, abrogates G2/M arrest and promotes DNA re-replication. A, Cells were treated with MLN4924, SCH 900776 or the combination of MLN4924 and SCH 900776 at indicated concentrations for 24 hours, followed by IB analysis. B, synchronized Hup-T3 cells were treated with DMSO, MLN4924 or the combination of MLN4924 and SCH 900776, and collected at indicated time points, followed by FACS analysis for cell cycle profiling. C, asynchronized cells were treated with DMSO, MLN4924, SCH 900776 or the combination of MLN4924 and SCH 900776 for 24 hours, followed by FACS analysis. *, P<0.05; **, P>0.05; MLN, MLN4924; SCH, SCH 900776; DMSO, dimethyl sulfoxide; PI, propidium iodide. >4N represents cells having greater than tetraploid DNA content.
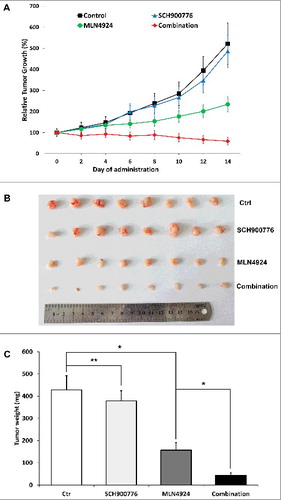
SCH 900776 increased the efficacy of MLN4924 in an in vivo xenograft tumor model
We assessed the efficacy of MLN4924, SCH 900776, and the combination therapy in vivo using the Panc-1 xenograft model. As shown in A-B, MLN4924 alone had a moderate inhibitory effect on tumor growth in nude mice, and SCH 900776 alone had almost no impact on tumor growth, while the combination of MLN4924 and SCH 900776 significantly inhibited tumor growth. At the end of administration, the TGI of SCH 900776 group, MLN4924 group, and the combination therapy group was 6.53%, 55.09%, 88.68%, respectively, with differences between each group statistically significant (P<0.05). The average tumor weight of MLN4924 group, SCH 900776 group and the combination therapy group was 428.4 mg, 379.1 mg, 157.4 mg, 43.9 mg, respectively, with difference between the MLN4924 group and the combination therapy group statistically significant (P<0.05) (C). Meanwhile all the treatment was well tolerated by the animals with a minimal loss of body weight. These results are consistent with in vitro studies. Taken together, we showed that SCH 900776 enhances the antitumor efficacy of MLN4924 on pancreatic cancer, likely by abrogating the G2/M arrest and reinforce DNA re-replication.
Figure 5. SCH 900776 increased the efficacy of MLN4924 in Panc-1 in vivo xenograft tumor model. 1 × 106 Panc-1 cells were inoculated subcutaneously in the left flank of nude mice. When the tumor size reached 100 mm3, the mice were randomized into the following 4 groups (10 animals/group) and treated with (1) 20% HPBCD, (2) MLN4924 (30 mg/kg, s.c.), (3) SCH 900776 (8 mg/kg, i.p.), (4) MLN4924 combined with SCH 900776 at the preceding dosage, on a five days on and two days off schedule for two weeks. The tumor growth curve was plotted (A) and at the end of the study, tumors were resected (B) and weighed (C). *, P<0.05; **, P>0.05.
NEDD8-activating enzyme level was elevated in pancreatic cancer and was irrelevant with prognosis
The clinicopathological features of 90 pancreatic cancer patients are listed in . As a reference, A shows the representative staining of normal pancreas tissue and pancreatic cancer tissue of different NAE-expression levels. The NAE expression levels of normal pancreas tissue and pancreatic cancer tissue of 90 patients are listed in . The percentage of high NAE-expression rate of pancreatic cancer was higher than normal pancreas tissue (54.4% vs. 21.1%, p<0.05). In general, pancreatic cancer tissue has higher NAE-expression levels than normal pancreas tissue (p<0.05). We then compared the prognosis of patients with pancreatic cancer of different NAE-expression levels, and found no significant difference between the survival of patients with pancreatic cancer of low or medium NAE expression and of high NAE expression (median survival, 15.9 vs 14.6 months, p>0.05) (B).
Table 1. Clinicopathological features of 90 pancreatic cancer patients.
Figure 6. NEDD8-activating enzyme levels of pancreatic cancer were irrelevant with prognosis. A, representative staining of normal pancreas tissue and pancreatic cancer tissue of different NAE-expression levels. B, the survival curves of patients with pancreatic cancer of low or medium NAE expression and of high NAE expression. NAE, NEDD8-activating enzyme.
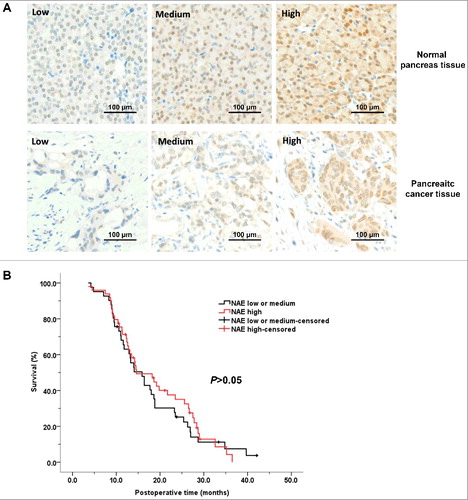
Table 2. The NAE expression levels of normal pancreas tissue and pancreatic cancer tissue of 90 patients.
Discussion
The dismal prognosis of pancreatic cancer requires more efficient therapeutic agents. As a novel small molecule inhibitor of NAE, MLN4924 drew our attention because of its wide range of antitumor activities and good tolerance in vivo [Citation19]. Previous studies have focused more on the sensitizing effects of MLN4924 to the existing chemotherapy agents [Citation32,Citation33] or radiation therapy [Citation22,Citation29]. But as shown in previous studies [Citation22,Citation27] and in ours, MLN4924 alone causes DNA damage on cancer cells, and inspired by the success of proteasome inhibitor Bortezomib [Citation34], we wondered if MLN4924 could act as a primary cytotoxic agent toward pancreatic cancer. Meanwhile, because the DNA damage response elicited by MLN4924 leaded to checkpoint activation and cell cycle arrest, we hypothesized that adding a Chk1 inhibitor would enhance the antitumor efficacy. SCH 900776 was a potent and functionally selective inhibitor of Chk1 currently in clinical development. In this study, we evaluated the effects of the combination therapy of MLN4924 and SCH 900776 in vitro and in vivo.
Consistent with the previous study [Citation22], we found that MLN4924 alone was a potent tumor growth inhibitor, and SCH 900776 alone didn't affect the survival and proliferation of pancreatic cancer cells until at a fairly high concentration. For the first time, we showed here that SCH 900776 enhanced the tumor killing of MLN4924, both on cultured pancreatic cancer cells and on an in vivo xenograft tumor model, using two different pancreatic cell lines. Importantly, no obvious toxicity was observed in all the treatment groups. Because of its safety and efficacy towards a variety of malignancies, MLN4924 has been advanced to several phase I and one phase II clinical trials for solid tumors and hematologic malignancies [Citation35,Citation36]. While there were less trials of SCH 900776, multiple other Chk1 inhibitors were being tested in the clinical setting [Citation37]. Our results provided the first preclinical evidence for further clinical studies of the combination therapy of MLN4924 and SCH 900776 or other Chk1 inhibitors on pancreatic cancer.
It is reported that MLN4924 treatment caused DNA damage as elevated level of phosphorylated Chk1 or Chk2 was observed [Citation19,Citation27]. Here by measuring the formation of 53BP1 foci, we offered the direct evidence that MLN4924 caused DSB, since 53BP1 is exclusively involved in the DSB repair pathway [Citation38]. Also, after MLN4924 treatment we detected cells experiencing G2/M arrest, which is a consequence of DNA damage response, and cells having greater than tetraploid DNA content. These results agree with the DNA rereplication hypothesis that MLN4924 treatment causes the accumulation of DNA replication licensing protein CDT1 [Citation38], a substance of CRLs [Citation39], which leads to more than one initiation of DNA replication per cell cycle. Rereplication results in genomic instability and subsequent DNA damage. Combining SCH 900776 with MLN4924, we found higher levels of phosphorylated Chk1, phosphorylated ATM, and γ-H2AX in treated cells, indicating more severe DNA damage was generated. This could be explained by the abrogation of G2/M arrest induced by SCH 900776, as shown in our study, allowing a damaged cell passing through S and G2 into mitosis before DNA damage was fully repaired [Citation37]. Moreover, cells treated with MLN4924 plus SCH 900776 had a higher population with DNA contents greater than 4N compared with MLN4924 alone, suggesting DNA rereplication was promoted by Chk1 inhibitor. This phenomenon may be related with the role of Chk1 in the stabilization of stalled replication forks [Citation40]. We found that the NAE level was elevated in pancreatic cancer compared with normal pancreas, maybe because tumor cells have a more active UPS system as they are metabolically more active [Citation41]. But the NAE level of pancreatic cancer was irrelevant with prognosis, so the biological significance of this upregulation remained obscure.
In summary, our study revealed that MLN4924 inhibited the proliferation, induced DNA damage and caused cell cycle arrest of pancreatic cancer cells. SCH 900776 alone did not inhibit the proliferation or cause DNA damage, but combined with MLN4924, SCH 900776 enhanced the antitumor activity and the DNA damage effects of MLN4924, likely by abrogating the G2/M arrest and reinforcing DNA re-replication. The drug efficacy study on mouse model further confirmed the antitumor activity of MLN4924 on pancreatic cancer, and SCH 900776 acted a sensitizer of MLN4924. Our study, therefore, provides the rationale and preclinical evidence of combining SCH 900776 or other Chk1 inhibitors with MLN4924 as a novel approach to treat pancreatic cancer.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suppl2017CC7689R-f07-z-4c.tif
Download TIFF Image (2.8 MB)Acknowledgments
We thank Dr. Larry M. Karnitz for providing the anti-Chk1 antibody and Ms. Haixia Zeng for help on the establishment of tissue microarrays.
Additional information
Funding
Related Research Data
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA A Cancer J Clin. 2016;66(1):7. doi:10.3322/caac.21332.
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338. PMID:26808342
- Manuel H. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. doi:10.1056/NEJMra0901557. PMID:20427809
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi:10.1056/NEJMoa1011923. PMID:21561347
- Aboualfa GK. Increased survival in pancreatic cancer with nab-Paclitaxel plus Gemcitabine. N Engl J Med. 2013;369(18):1691–703.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi:10.1056/NEJMoa050753. PMID:16014882
- Moscovici M. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857. PMID:18650514
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with Lambrolizumab (Anti–PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi:10.1056/NEJMoa1305133. PMID:23724846
- Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(351):337–345. doi:10.1056/NEJMoa033025. PMID:15269313
- Piccartgebhart MJ, Procter M, Leylandjones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. doi:10.1056/NEJMoa052306. PMID:16236737
- Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. doi:10.1200/JCO.2006.07.9525. PMID:17452677
- Herrmann J, Lerman LO, Lerman A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ Res. 2007;100(9):1276. doi:10.1161/01.RES.0000264500.11888.f0. PMID:17495234
- Kane RC, Farrell AT, Sridhara R, et al. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res. 2006;12(10):2955–2960. doi:10.1158/1078-0432.CCR-06-0170. PMID:16707588
- Kane RC, Dagher R, Farrell A, et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13(1):5291–5294. doi:10.1158/1078-0432.CCR-07-0871. PMID:17875757
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi:10.1152/physrev.00027.2001. PMID:11917093
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi:10.1038/nrm1547. PMID:15688063
- Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12(4):220. doi:10.1186/gb-2011-12-4-220. PMID:21554755
- Pan ZQ, Kentsis A, Dias DC, et al. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23(11):1985–97. doi:10.1038/sj.onc.1207414. PMID:15021886
- Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi:10.1038/nature07884. PMID:19360080
- Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic In Situ. Mol Cell. 2010;37(1):102. doi:10.1016/j.molcel.2009.12.024. PMID:20129059
- Milhollen MA, Narayanan U, Soucy TA, et al. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71(8):3042–3051. doi:10.1158/0008-5472.CAN-10-2122. PMID:21487042
- Wei D, Li H, Yu J, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282–293. doi:10.1158/0008-5472.CAN-11-2866. PMID:22072567
- Godbersen JC, Humphries LA, Danilova OV, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NFκB activation and induces apoptosis in chronic lymphocytic leukemia B-cells. Clin Cancer Res. 2014;20(6):1576. doi:10.1158/1078-0432.CCR-13-0987. PMID:24634471
- Milhollen MA, Traore TaDJ, Thomas MP, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116(9):1515–1523. doi:10.1182/blood-2010-03-272567. PMID:20525923
- Luo Z, Yu G, Lee HW, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72(13):3360–3371. doi:10.1158/0008-5472.CAN-12-0388. PMID:22562464
- Gu Y, Kaufman JL, Bernal L, et al. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. 2014;123(21):3269–3276. doi:10.1182/blood-2013-08-521914. PMID:24713927
- Lin JJ, Milhollen MA, Smith PG, et al. NEDD8-Targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S Phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. 2010;70(24):10310–10320.
- Mackintosh C, García-Domínguez DJ, Ordóñez JL, et al. WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene. 2013;32(11):1441–1451. doi:10.1038/onc.2012.153. PMID:22641220
- Yang D, Tan M, Wang G, et al. The p21-Dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS One. 2012;7(3):e34079. doi:10.1371/journal.pone.0034079. PMID:22457814
- Montano R, Chung I, Garner KM, et al. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11(2):427–438. doi:10.1158/1535-7163.MCT-11-0406. PMID:22203733
- Dima SO, Tanase C, Albulescu R, et al. An exploratory study of inflammatory cytokines as prognostic biomarkers in patients with ductal pancreatic adenocarcinoma. Pancreas. 2012;41(7):1001–1007. doi:10.1097/MPA.0b013e3182546e13. PMID:22722257
- Garcia K, Blank JL, Bouck DC, et al. Nedd8-activating enzyme inhibitor MLN4924 provides synergy with Mitomycin C through interactions with ATR, BRCA1/BRCA2 and chromatin dynamics pathways. Mol Cancer Ther. 2014;13:1625–1635. doi:10.1158/1535-7163.MCT-13-0634. PMID:24672057
- Nawrocki ST, Kelly KR, Smith PG, et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res. 2013;19(13):3577. doi:10.1158/1078-0432.CCR-12-3212. PMID:23633453
- Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. doi:10.1056/NEJMoa030288. PMID:12826635
- Oladghaffari M, Islamian JP, Baradaran B, et al. MLN4924 therapy as a novel approach in cancer treatment modalities. J Chemother. 2016;28(2):74. doi:10.1179/1973947815Y.0000000066. PMID:26292710
- Malhab LJB, Descamps S, Delaval B, et al. The use of the NEDD8 inhibitor MLN4924 (Pevonedistat) in a cyclotherapy approach to protect wild-type p53 cells from MLN4924 induced toxicity. Sci Rep. 2016;6:37775. doi:10.1038/srep37775. PMID:27901050
- Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76(3):358–369. doi:10.1111/bcp.12139. PMID:23593991
- Sulli G, Di Micco R, di Fagagna F d'Adda. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12(10):709–720. doi:10.1038/nrc3344. PMID:22952011
- Nishitani H, Sugimoto N, Roukos V, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. Embo J. 2006;25(5):1126–1136. doi:10.1038/sj.emboj.7601002. PMID:16482215
- Scorah J, Mcgowan CH. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle. 2009;8(7):1036–1043. doi:10.4161/cc.8.7.8040. PMID:19270516
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013. PMID:21376230