
ABSTRACT
Polo-like kinase 1 (Plk1) is an instrumental kinase that modulates many aspects of the cell cycle. Previous investigations have indicated that Plk1 is a target of the DNA damage response, and Plk1 inhibition is dependent on ATM/ATR and Chk1. But the exact mechanism remains elusive. In a proteomic screen to identify Chk1-interacting proteins, we found that myosin phosphatase targeting protein 1 (MYPT1) was present in the immunocomplex. MYPT1 is phosphorylated by CDK1, thus recruiting protein phosphatase 1β (PP1cβ) to dephosphorylate and inactivate Plk1. Here we identified that Chk1 directly interacts with MYPT1 and preferentially phosphorylates MYPT1 at Ser20, which is essential for MYPT1-PP1cβ interaction and subsequent Plk1 dephosphorylation. Phosphorylation of Ser20 is abolished during mitotic damage when Chk1 is inhibited. The degradation of MYPT1 is also regulated by Chk1 phosphorylation. Our results thus unveil the underlying machinery that attenuates Plk1 activity during mitotic damage through Chk1-induced phosphorylation of MYPT1.
Abbreviations
| Plk1 | = | Polo-like kinase 1 |
| Noc | = | nocodazole |
| IP | = | immunoprecipitation |
| IB | = | immunoblotting |
| Chk1 | = | checkpoint kinase 1 |
| MYPT1 | = | myosin phosphatase targeting protein 1 |
| IVK | = | in vitro kinase |
| PP1cβ | = | protein phosphatase 1β |
| LC-MS/MS | = | liquid chromatography-tandem mass spectrometry |
| PBD | = | polo-binding domain |
| ATM | = | ataxia telangiectasia mutated |
| ATR | = | ataxia telangiectasia and Rad3 related |
| coIP | = | coimmunoprecipitation |
| WT | = | wild type |
Introduction
Plk1 is a vital kinase that orchestrates many cell cycle events, including DNA replication, centrosome duplication, mitotic entry, spindle assembly, chromosome segregation and eventual cytokinesis [Citation1]. It comprises an N-terminal kinase domain and a C-terminal polo-box binding domain (PBD), which frequently mediates its binding with substrates via a pSer/Thr-Pro motif [Citation1]. The T-loop of Plk1 harbors phospho-T210, a prerequisite for Plk1 activation. The central role of Plk1 in the cell cycle is subject to multilayers of regulation. For instance, the kinase activity of Plk1 starts to increase at the G2/M phase, maximizing at mitosis, which is regulated by Aurora A-dependent phosphorylation at T210 [Citation2,Citation3], facilitated by BORA. During DNA damage response (DDR), the recruitment of Aurora A by BORA is perturbed, resulting in loss of pT210 and Plk1 inactivation [Citation4]. Plk1 is also inactivated in DDR by a pathway dependent on ataxia telangiectasia mutated (ATM) / ataxia telangiectasia and Rad3 related (ATR) [Citation5,Citation6] and Chk1 [Citation7] and degraded by the E3 ligase Anaphase Promoting Complex/Cyclosome (APC/C) [Citation8]. The substrates of Plk1 in DDR are emerging. Plk1 phosphorylates Mre11 at Ser649, inhibiting the recruitment of the Mre11/Rad50/Nbs1 (MRN) complex to damaged sites [Citation9]. Moreover, Plk1 antagonizes p53 during DDR, via phosphorylating Numb, which is also a cell-fate determinant in stem/progenitor cells [Citation10].
The exact mechanism remains elusive as to how Plk1 is switched off by Chk1 [Citation7]. In response to DDR, Chk1 is activated by either ATM or ATR [Citation11,Citation12], then targets myriad substrates that mediate downstream events. In the absence of DNA damage, Chk1 also plays pivotal roles in DNA replication, intra-S phase checkpoints, G2/M checkpoints and spindle-assembly checkpoints [Citation13,Citation14]. Therefore, Chk1 is currently one of the most promising therapeutic targets for numerous cancers [Citation15].
In spite of its emerging significance in anticancer research, many substrates of Chk1 are yet to be identified. We sought to study the Chk1 network by identifying more Chk1-interacting proteins. In the immunocomplexes of Chk1, we identified myosin phosphatase targeting protein 1 (MYPT1, or PPP1R12A), the targeting subunit of protein phosphatase 1cβ (PP1cβ) [Citation16]. Human PP1 is encoded by three related genes, and includes three isoforms: α, β/δ and γ. MYPT1 contains ∼ 1, 000 amino acids with a mass of ∼ 110 kDa and specifically binds with PP1cβ/δ. It has a conserved N-terminal region of seven ankyrin repeats as well as a characteristic PP1c-binding motif (35KVKF38) [Citation17]. Previous investigations have implicated PP1 regulators in DDR, such as Repo-Man (binds with PP1cγ) and PNUTS (binds all three isoforms) [Citation18]. But MYPT1 has never been suspected.
MYPT1's major role in mitosis has been proposed to regulate the activity of Plk1. The dephosphorylation of Plk1 at pT210 by MYPT1 is regulated multifacetedly: MYPT1 is phosphorylated by Cdk1 at Ser437 during mitosis to target PP1cβ to Plk1 [Citation19]; MYPT1 is phosphorylated by LATS1/WARTS at Ser445 to antagonize Plk1 [Citation20]; MYPT1 activity is also promoted by another phosphatase, optineurin, forming a negative feedback loop to modulate Plk1 activity [Citation21]. Here we identify that Chk1 interacts with and phosphorylates MYPT1 at Ser20, which is essential for MYPT1-PP1cβ binding. We propose that Chk1-dependent phosphorylation of MYPT1 promotes the MYPT1-PP1cβ phosphatase activity towards Plk1.
Results
Identification of MYPT1 as a novel target for Chk1
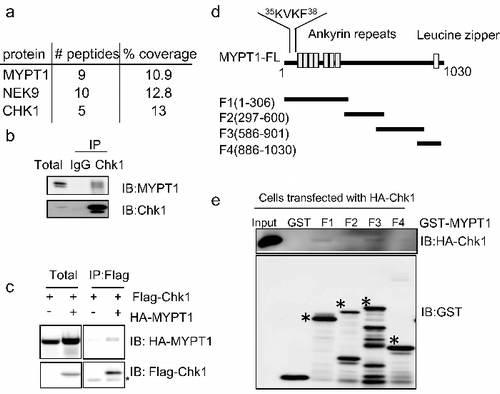
In an effort to identify Chk1-interacting proteins, we performed immunoprecipitation (IP) assays with anti-Chk1 antibodies followed by mass spectrometry (MS) analysis as previously described [Citation22]. The assay identified not only Chk1 itself, but also known proteins that interact with Chk1 (such as NEK9 [Citation23]), as well as many novel interacting proteins, among which was MYPT1 (A). To validate the potential association between Chk1 and MYPT1, HeLa cell extracts were subject to coIP experiments using anti-Chk1 antibodies, and MYPT1 was detected in the immunoprecipitates (B). We also co-transfected Flag-Chk1 and HA-MYPT1 into HeLa cells and an interaction was also observed between the overproduced proteins (C), excluding the possibility that the interaction was an artifact due to antibody non-specificity. To determine the regions of MYPT1 required for its interaction with Chk1, we constructed GST-tagged wild-type (WT) MYPT1 and a series of MYPT1 fragments (F1-F4) (D) to carry out coIP experiments with full-length (FL) HA-tagged Chk1. As shown in E, Chk1 preferentially interacted with F1 (aa. 1–306) and F3 (aa. 586–901) regions of MYPT1. Taken together, these data suggest that Chk1 associates with MYPT1.
Figure 1. Interaction between Chk1 and MYPT1. (A) Chk1 was immunoprecipitated from HeLa cells, and samples were analyzed by SDS-PAGE with Coomassie blue staining. Chk1-interacting proteins were identified by mass spectrometry. Shown in the table are the proteins identified in the immunocomplex. The number of peptides and peptide coverage pertentage were indicated. (B) Chk1 immunoprecipitates were blotted with anti-Chk1 and anti-MYPT1 antibodies.(C) HeLa cells transfected with vector or HA-MYPT1, and Flag-Chk1 constructs were subjected to IP and IB assays using the antibodies indicated. Asterisks indicate IgG. (D) Domain structures of MYPT1. The PP1 binding motif, the ankyrin repeats, and the Leucine zipper domain, and the boundaries of MYPT1 fragments used in this study are indicated. (E) Bacterially expressed MYPT1 fragments were incubated with lysates from HeLa cells transfected with HA-Chk1. Asterisks indicate the corresponding bands.
Chk1-dependent phosphorylation of MYPT1 at S20
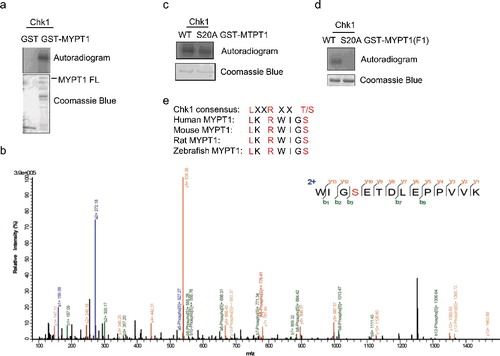
To investigate whether Chk1 regulates MYPT1 through phosphorylation, we used in vitro kinase (IVK) assays with recombinant Chk1 and MYPT1. Chk1 phosphorylates MYPT1 efficiently (A). The IVK products were then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), and Ser20 was identified as the most abundant phosphorylation site (B). When Ser20 was mutated to Ala in MYPT1 and used in the IVK assay, the phosphorylation signal was reduced about ∼20% in FL MYPT1-S20A (C). We then constructed S20A in MYPT1-F1, and the phosphorylation signal was completely abolished in MYPT1-F1-S20A (D). The amino acid sequence surrounding the Ser20 residue matched a minimum consensus for Chk1-dependent phosphorylation, and it is also conserved among MYPT1 homologues (E). These results suggest that Ser20 is a major phosphorylation site in F1. Since MYPT1 is a relative large protein of 1,030 amino acids, there are probably other Chk1-dependent phosphorylation sites on MYPT1.
Figure 2. Ser20 is identified as one of the Chk1-dependent phosphorylation sites. (A) Recombinant Chk1 and MYPT1 were incubated in kinase buffers containing 32P-ATP, and an IVK assay was carried out. Coomassie blue staining showed input MYPT1 proteins and autoradiography showed phosphorylated GST-MYPT1. (B) LC-MS/MS analysis identified Ser20 of MYPT1 as one of the sites phosphorylated by Chk1 in vitro. From this collision-induced dissociation spectrum, a phosphorylated peptide WIG(pS)ETDLEPPVVK of MYPT1 was identified following incubation with Chk1 in an IVK reaction. “b” and “y” ion series represent fragment ions containing the N- and C-termini of the peptide, respectively. (C-D) S20A mutant was constructed in GST-MYPT1-FL (C) and -F1 (D) fragment, and IVK assays were carried out using WT and S20A of GST-MYPT1-FL or F1 proteins. (E) A comparison between Chk1 phosphorylation consensus sites and the adjacent amino acids of MYPT1 Ser20.
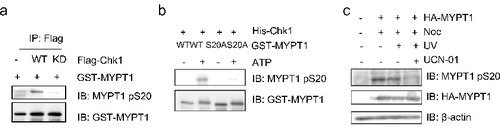
To validate that MYPT1 is an in vivo substrate of Chk1, we raised a polyclonal phospho-specific antibody, MYPT1-pS20, as described in Experimental procedures. Using immunoprecipitated Chk1 in the IP-kinase assay, MYPT1-pS20 was readily detectable with WT-Chk1, but not with the Chk1-kinase dead (KD), suggesting that MYPT1-pS20 was dependent upon Chk1 kinase activity (A). We then used commercially available recombinant Chk1 and measured the level of pS20 in an IVK assay with MYPT1-WT or S20A. As demonstrated in B, Chk1 efficiently phosphorylated MYPT1, but not MYPT1-S20A. We noted that GST-MYPT1-S20A displayed distinct migration patterns in the presence or absence of ATP, suggesting that there are other phosphorylation events involved. Then we tested whether MYPT1 is phosphorylated at pS20 in vivo. HeLa cells were treated with Nocodazole (Noc), Noc plus UV, or Noc plus UV plus UCN-01 (an inhibitor of Chk1). Immonoblotting with pS20 antibodies indicated that MYPT1 is phosphorylated at pS20 upon Noc treatment, and upon Noc plus UV treatment. But the band was significantly diminished upon UCN-01 treatment (C), suggesting that pS20 is dependent on Chk1. Collectively, the results indicate that pS20 of MYPT1 depends on the kinase activity of Chk1.
Figure 3. Ser20 of MYPT1 is phosphorylate by Chk1. (A) HeLa cells were transfected with Flag-Chk1 or Flag-Chk1-kinase dead (KD), and then IPed with anti-Flag antibodies. The immunoprecipitates were used in an IP-kinase assay using recombinant GST-MYPT1 as substrates. The reaction was then terminated and blotted with antibodies indicated. (B) IVK assays using recombinant His-Chk1 and GST-MYPT1 or GST-MYPT1-S20A as substrates. The products were then blotted with pS20 antibodies. (C) Ser20 of MYPT1 was phosphorylated in vivo. HeLa cells transfected with HA-MYPT1 were treated with Noc, Noc plus UV, Noc plus UV plus UCN-01 (an inhibitor of Chk1). Lysates from these cells were blotted with pS20 antibodies.
pS20 promotes degradation of MYPT1
Since the proteasome-dependent degradation of MYPT1 is mediated by the SIAH2 E3 ligase [Citation24], we examined whether Chk1-induced phosphorylation of Ser20 affected the stability of MYPT1. Cells were transfected with Flag-Chk1 and HA-Ub, and then IPed with anti-MYPT1 antibodies. The ubiquitination levels of MYPT1 were elevated compared with controls (A), suggesting that Chk1 overproduction increases MYPT1 ubiquitition. To differentiate whether it is due to the protein level or the kinase activity of Chk1, we transfected WT or KD of Chk1 into cells. The ubiquitination levels of MYPT1 in the cells transfected with Chk1-KD were markedly down-regulated (B), indicating that the kinase activity of Chk1 is essential. To confirm this, we also utilized UCN-01 to inhibit Chk1. Again, Chk1 inhibition decreased ubiquitination levels of MYPT1 (C).
Figure 4. pS20 promotes the degradation of MYPT1. (A) HeLa cells transfected with Flag-Chk1 and HA-Ub were treated with MG132 or left untreated, IPed with anti-MYPT1 antibodies and IBed with the antibodies indicated. (B) Cells were transfected with vector, WT or KD of Flag-Chk1 and HA-Ub plasmids, then analyzed as in (A). (C) Cells were treated with UCN-01 or left untreated, then analyzed as in A. (D) Cells were transfected with HA-MYPT1, treated with UCN-01 or left untreated, then incubated with CHX for the time indicated. (E) Quantitation of the results in (D). (F) Cells were transfected with WT or S20A of HA-MYPT1, then incubated with CHX. (G) Quantitation of the results in (F).
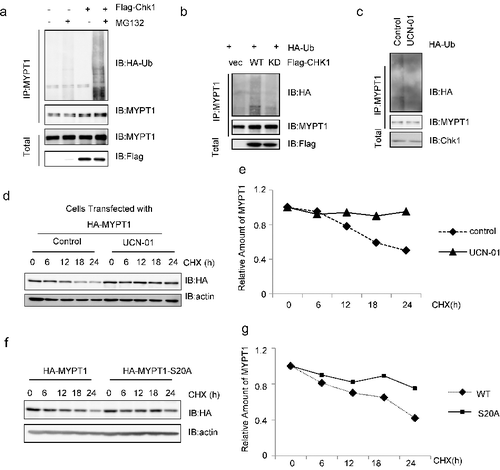
Since ubiquitination often leads to degradation, we adopted cycloheximide (CHX), a protein synthesis inhibitor, to assess the protein stability of MYPT1. As shown in D-E, UCN-01 treatment increased the stability of MYPT1. Moreover, when WT or S20A of MYPT1 were transfected into cells and treated with CHX, MYPT1-S20A manifested longer half-life (F-G). In sum, Chk1 promotes the ubiquitination and subsequent degradation of MYPT1, probably through phosphorylating S20.
S20 phosphorylation is essential for the interaction between MYPT1 and PP1cβ
As reported previously, MYPT1 contains a PP1c binding motif (KVSF) at its N-terminus [Citation17], adjoining to the site of S20 (D). We speculated interplay between phosphorylation of S20 and PP1cβ binding. Indeed, when we transfected MYPT1-WT and S20A into HeLa cells, the interaction between MYPT1-S20A and PP1cβ was completely abolished, as examined by coIP assays (A). Then we tested the interaction by pulldown assays (B). Recombinant GST-MYPT1-WT or S20A proteins were used to pull down transfected HA-PP1cβ. We observed that GST-MYPT1-S20A significantly compromised the interaction with PP1cβ (B).
Figure 5. pS20 is essential for the interaction between MYPT1 and PP1cβ. (A) HeLa cells were transfected with HA-MYPT1-WT or S20A plasmids, then subject to IP and IB analysis as indicated. (B) Recombinant GST-MYPT1 and S20A proteins were used to pulldown transfected HA- PP1cβ. (C) HeLa cells were transfected with HA-MYPT1-WT or S20A plasmids and treated with Noc, then subject to IP with anti-HA antibodies, followed by IP-phosphatase assays using active Plk1 as substrates. The products were then blotted with anti-Plk1-pT210 antibodies. (D) A proposed model of how Chk1 inhibits Plk1 through MYPT1. Chk1 phosphorylates MYPT1 at Ser20 to promote the interaction between MYPT1 and PP1cβ. MYPT1 then targets PP1cβ to Plk1 to dephosphorylate Plk1 at pT210, and possibly other proteins.
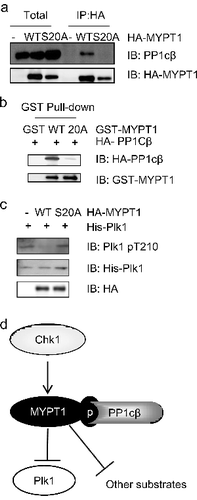
We reasoned that since MYPT1-S20A could not target PP1cβ to its substrate, then Plk1 may not be dephosphorylated by MYPT1-S20A. We thus assessed this possibility by an IP-phosphatase assay. HeLa cells were transfected with HA-MYPT1-WT or S20A plasmids, and then treated with Noc. As reported previously, activated Cdk1 will phosphorylate MYPT1 at S473 to target MYPT1 to dephosphorylate Plk1 [Citation19]. The cell extracts were IPed with anti-HA antibodies, and incubated with active Plk1 (C). Plk1 was efficiently dephosphorylated by MYPT1-WT, as previously described [Citation19], but not by MYPT1-S20A (C), suggesting that phosphorylation at S20 plays a pivotal role in promoting the MYPT1-PP1cβ activity towards Plk1.
Discussion
Our results suggest that MYPT1 is a substrate of Chk1, and Chk1 phosphorylates MYPT1 at Ser20. There are probable other phosphorylation sites, evidenced by the results below: Chk1 still phosphorylates GST-MYPT1-S20A in IVK assays, albeit at a lesser degree; S20 is not the sole site identified in the MS analysis; GST-MYPT1-S20A still displays distinct band shift upon Chk1 incubation, indicative of phosphorylation (B). Nevertheless, our results indicate that Ser20 is the most abundant phosphorylation site identified in MS, and Ser20 is phosphorylated by Chk1 in vivo and in vitro.
Phosphorylation of Ser20 was identified recently in a proteomic study of MYPT1 [Citation25], but its functional role was left unattended. Our results also indicate that pS20 is pivotal for MYPT1-PP1cβ binding, and therefore PP1cβ activity towards its substrates (D). First, MYPT1-S20A fails to coIP with PP1cβ (A). Second, MYPT1-S20A fails to pulldown PP1cβ (B). Lastly, one of MYPT1-PP1cβ's substrate, Plk1, fails to be dephosphorylated by MYPT1-S20A immunoprecipitates (C).
Here we identified that phosphorylation of MYPT1 by Chk1 is subject to ubiquitin-mediated degradation. A query into TCGA [Citation26,Citation27] revealed that MYPT1 is overexpressed in a variety of cancer types, including neuroendocrine prostate cancer (NePC), breast, prostate and sarcoma, and deleted in pancreas cancer, suggesting that the exact level of MYPT1 is essential to exert its function. Chk1-mediated crosstalk between phosphorylation and ubiquitination is quite common, for instance, Chk1 phosphorylates spleen tyrosine kinase (SYK) at Ser295 for degradation [Citation28], and Chk1 phosphorylates O-Linked β-N-Acetylglucosamine Transferase (OGT) at Ser20 for stabilization [Citation22]. This pattern was also observed for other kinases: Cdc25A is phosphorylated by glycogen synthase kinase -3β (GSK3β) for degradation [Citation29]. But the remaining question is, why does Chk1 degrade MYPT1 while at the same time promote the interaction between MYPT1 and PP1cβ? This seeming paradox might be rooted in Plk1, whose activity is downregulated during DDR [Citation5], but not completely abolished [Citation9,Citation10]. We reasoned that Chk1 might modulate an intricate balance between MYPT1 protein level and MYPT1-PP 1cβ activity, so that the exact amount of Plk1 is active during DDR.
It is highly likely that other substrates of PP1cβ will also be affected by MYPT1-S20. Of the many proteins identified to interact with MYPT1 [Citation30], quite a few are associated with human diseases, for instance, the Tau protein (associated with the Alzheimer's), the retinoblastoma protein, and of particular interest, is the Merlin protein [Citation30]. Merlin is a tumor suppressor protein that regulates the Hippo signaling pathway, a tumor suppressor pathway inactivated in many cancers. It will be of interest to examine whether Ser20 phosphorylation modulates the Hippo signaling pathway. The S20 site we identified here thus adds to the repertoire of MYPT1-PP1cβ regulation, and may cue in further therapeutic interventions for human diseases.
Experimental procedures
Cell culture, antibodies and plasmids
HeLa and HEK293 cells were purchased from ATCC. Anti-MYPT1 antibodies, MYPT1 plasmids were described before [Citation31]. Plk1-pT210 antibodies were from BD (cat. 558400). Antibodies against MYPT1 phospho-Ser20 (pS20-Ab) were raised in rabbits using the sequence of KRWIG(pS)ETDLC and manufactured by Beijing B&M Biotech Co., Ltd. MYPT1-S20A mutants were generated using specific primers (sequences available upon request) following the manufacturer's instructions (QuickChange II, Stratagene).
Transfections
HeLa cells were transfected twice with a 24-h interval using Oligofectamine (Invitrogen) according to the manufacturer's instructions. Transfectants were used for further experiments 24 hrs after the second transfection.
For plasmid transfection, cells were seeded at 50–60% confluence/10 cm2 petri dish and transfected with 7.5 μg of plasmid DNA using FuGene 6 according to the manufacturer's instructions for immunoprecipitation (IP) experiments.
IP and Immunoblotting (IB)
IP and IB experiments were performed as described before [Citation32]. Where indicated, UCN-01 was used at 30 nM for 2 hr. Cells were treated with UV at 365 nm for 2 min. The following primary antibodies were used for immunoblotting: anti-MYPT1, anti-Chk1, anti-β-actin, anti-HA, anti-Myc and anti-FLAG M2 (Sigma). Peroxidase-conjugated secondary antibodies were from JacksonImmuno Research. Blotted proteins were visualized using the ECL detection system (Amersham). Signals were detected by a LAS-4000, and analyzed using Multi Gauge (Fujifilm).
In vitro kinase (IVK) assay
Chk1 IVK assay was performed as previously described. Briefly, recombinant Chk1 kinase was purchased from R & D systems (Cat. # 1630-KS), incubated with purified GST-MYPT1 with 1 M HEPES (pH 7.4), 1 M MgCl2, 1 M Dithiothreitol, 0.1 M Na3VO4, 0.1 mM ATP or 1 μCi of γ-[32P]ATP. After 20 min at 30°C, reactions were stopped by the sample buffer. Protein samples were separated by SDS-PAGE and phosphate incorporation was determined by Phosphor Imager. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis were performed as described [Citation33].
IP-phosphatase assays
IP-phosphatase assays were performed as described previously [Citation20]. HeLa cells were transfected with wild-type (WT) HA-MYPT1 or S20A treated with nocodazole (Noc) (100 ng/ml for 16 hrs), then subjected to IP with anti-HA antibodies. Immunoprecipitates were washed in lysis buffers and preincubated at 4°C for 10 min with PP1 assay buffers (20 mM Tris-HCl, pH 7.4, 1% Triton X-100, 250 mM sucrose, 1 mM MnCl2, and 0.1% β-mercaptoethanol). Next, 0.2 μg of active Plk1 (Carna Biosciences, Inc.) was added to the reaction solution to a final volume of 20 μl. Phosphatase reactions were performed at 30°C for 30 min and terminated, then followed by Western analysis.
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgements
J. Li was supported by Beijing Nova Program Interdisciplinary Cooperation Project (Z161100004916042) and CNU Interdisciplinary Project. X.X. was supported by NSFC funds (31530016 and 31761133012) the National Key R&D Program of China projects 2015CB910601 and 2017YFA0503900, and Shenzhen Science and Technology Innovation Commission (JCYJ20170412112009742).
Additional information
Funding
References
- Zitouni S, Nabais C, Jana SC, et al. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15:433–452. doi:10.1038/nrm3819. PMID:24954208
- Seki A, Coppinger JA, Jang CY, et al. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–1658. doi:10.1126/science.1157425. PMID:18566290
- Bruinsma W, Macurek L, Freire R, et al. Bora and Aurora-A continue to activate Plk1 in mitosis. J Cell Sci. 2014;127:801–811. doi:10.1242/jcs.137216. PMID:24338364
- Bruinsma W, Aprelia M, Garcia-Santisteban I, et al. Inhibition of Polo-like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene. 2017;36:1840–1848. doi:10.1038/onc.2016.347. PMID:27721411
- Smits VA, Klompmaker R, Arnaud L, et al. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–676. doi:10.1038/35023629. PMID:10980711
- van Vugt MA, Smits VA, Klompmaker R, et al. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem. 2001;276:41656–41660. doi:10.1074/jbc.M101831200. PMID:11514540
- Tang J, Erikson RL, Liu X. Checkpoint kinase 1 (Chk1) is required for mitotic progression through negative regulation of polo-like kinase 1 (Plk1). Proc Natl Acad Sci U S A. 2006;103:11964–11969. doi:10.1073/pnas.0604987103. PMID:16873548
- Bassermann F, Frescas D, Guardavaccaro D, et al. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi:10.1016/j.cell.2008.05.043. PMID:18662541
- Li Z, Li J, Kong Y, et al. Plk1 Phosphorylation of Mre11 Antagonizes the DNA Damage Response. Cancer Res. 2017;77:3169–3180. doi:10.1158/0008-5472.CAN-16-2787. PMID:28512243
- Shao C, Chien SJ, Farah E, et al. Plk1 phosphorylation of Numb leads to impaired DNA damage response. Oncogene. 2017. doi:10.1038/onc.2017.379.
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi:10.1128/MCB.21.13.4129-4139.2001. PMID:11390642
- Jazayeri A, Falck J, Lukas C, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi:10.1038/ncb1337. PMID:16327781
- Smith J, Tho LM, Xu N, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi:10.1016/B978-0-12-380888-2.00003-0. PMID:21034966
- Zachos G, Black EJ, Walker M, et al. Chk1 is required for spindle checkpoint function. Dev Cell. 2007;12:247–260. doi:10.1016/j.devcel.2007.01.003. PMID:17276342
- Carrassa L, Damia G. Unleashing Chk1 in cancer therapy. Cell Cycle. 2011;10:2121–2128. doi:10.4161/cc.10.13.16398. PMID:21610326
- Grassie ME, Moffat LD, Walsh MP, et al. The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys. 2011;510:147–159. doi:10.1016/j.abb.2011.01.018. PMID:21291858
- Ito M, Nakano T, Erdodi F, et al. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. 2004;259:197–209. doi:10.1023/B:MCBI.0000021373.14288.00. PMID:15124925
- Kuntziger T, Landsverk HB, Collas P, et al. Protein phosphatase 1 regulators in DNA damage signaling. Cell Cycle. 2011;10:1356–1362. doi:10.4161/cc.10.9.15442. PMID:21451260
- Yamashiro S, Yamakita Y, Totsukawa G, et al. Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev Cell. 2008;14:787–797. doi:10.1016/j.devcel.2008.02.013. PMID:18477460
- Chiyoda T, Sugiyama N, Shimizu T, et al. LATS1/WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J Cell Biol. 2012;197:625–641. doi:10.1083/jcb.201110110. PMID:22641346
- Kachaner D, Filipe J, Laplantine E, et al. Plk1-dependent phosphorylation of optineurin provides a negative feedback mechanism for mitotic progression. Mol Cell. 2012;45:553–566. doi:10.1016/j.molcel.2011.12.030. PMID:22365832
- Li Z, Li X, Nai S, et al. Checkpoint kinase 1-induced phosphorylation of O-linked beta-N-acetylglucosamine transferase regulates the intermediate filament network during cytokinesis. J Biol Chem. 2017;292(48):19548–19555. doi:10.1074/jbc.M117.811646.
- Smith SC, Petrova AV, Madden MZ, et al. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res. 2014;42:11517–11527. doi:10.1093/nar/gku840. PMID:25217585
- Twomey E, Li Y, Lei J, et al. Regulation of MYPT1 stability by the E3 ubiquitin ligase SIAH2. Exp Cell Res. 2010;316:68–77. doi:10.1016/j.yexcr.2009.09.001. PMID:19744480
- Chao A, Zhang X, Ma D, et al. Site-specific phosphorylation of protein phosphatase 1 regulatory subunit 12A stimulated or suppressed by insulin. J Proteomics. 2012;75:3342–3350. doi:10.1016/j.jprot.2012.03.043. PMID:22516431
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi:10.1126/scisignal.2004088. PMID:23550210
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi:10.1158/2159-8290.CD-12-0095. PMID:22588877
- Hong J, Hu K, Yuan Y, et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J Clin Invest. 2012;122:2165–2175. doi:10.1172/JCI61380. PMID:22585575
- Kang T, Wei Y, Honaker Y, et al. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi:10.1016/j.ccr.2007.12.002. PMID:18167338
- Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: Many roles in cell function. Biochem Biophys Res Commun. 2008;369:149–156. doi:10.1016/j.bbrc.2007.12.090. PMID:18155661
- Li J, Liu X, Liao J, et al. MYPT1 sustains centromeric cohesion and the spindle-assembly checkpoint. J Genet Genomics. 2013;40:575–578. doi:10.1016/j.jgg.2013.08.005. PMID:24238611
- Li J, Wang J, Hou W, et al. Phosphorylation of Ataxin-10 by polo-like kinase 1 is required for cytokinesis. Cell Cycle. 2011;10:2946–2958. doi:10.4161/cc.10.17.15922. PMID:21857149
- Tian J, Tian C, Ding Y, et al. Aurora B-dependent phosphorylation of Ataxin-10 promotes the interaction between Ataxin-10 and Plk1 in cytokinesis. Sci Rep. 2015;5:8360. doi:10.1038/srep08360. PMID:25666058