
ABSTRACT
The tumor suppressor p53, and the cyclin-dependent kinase inhibitor Ink4c, have been both implicated in spermatogenesis control. Both p53-/- and Ink4c-/- single knockout male mice are fertile, despite testicular hypertrophy, Leydig cell differentiation defect, and increased sperm count in Ink4c-/- males. To investigate their collaborative roles, we studied p53-/- Ink4c-/- dual knockout animals, and found that male p53-/- Ink4c-/- mice have profoundly reduced fertility. Dual knockout male mice show a marked decrease in sperm count, abnormal sperm morphology and motility, prolongation of spermatozoa proliferation and delay of meiosis entry, and accumulation of DNA damage. Genetic studies showed that the effects of p53 loss on fertility are independent of its downstream effector Cdkn1a. Absence of p53 also partially reverses the hyperplasia seen upon Ink4c loss, and normalizes the Leydig cell differentiation defect. These results implicate p53 in mitigating both the delayed entry into meiosis and the secondary apoptotic response that occur in the absence of Ink4c. We conclude that the cell cycle genes p53 and Ink4c collaborate in sperm cell development and differentiation, and may be important candidates to investigate in human male infertility conditions.
Introduction
The process of mammalian spermatogenesis has been extensively studied. It occurs in the seminiferous tubules of the testicles, proceeding from germ cells, the spermatogonia, at the periphery of the tubules towards the central lumen, during which time the spermatogonia undergo multiple mitotic divisions, gradually differentiating in preparation for meiosis. Primary spermatocytes undergo meiosis I, forming secondary spermatocytes which undergo meiosis II, producing haploid, round spermatids. These spermatids then undergo spermiogenesis, where they form a condensed spherical head with an acrosome, undergo elongation, and shed excess cytoplasm, producing mature spermatozoa that are released into the lumen [Citation1]. In mice, spermatogonia begin to differentiate around postnatal (P) day 8, or P8 [Citation2]. Primary spermatocytes are present by P10, and they undergo the first meiotic division at around P17-18 [Citation3]. Secondary spermatocytes divide again at around P20. Round spermatids are formed, undergo differentiation, and first mature sperm cells begin to form around P35, thus reaching puberty around the age of 6 weeks [Citation4].
Sertoli cells within the seminiferous tubules provide support for spermatocytes and spermatids, in addition to producing several growth factors and phagocytizing residual cytoplasm that is shed by spermatids during spermiogenesis. The other major supporting cell type is the Leydig cell, located between the tubules, the primary function of which is to produce testosterone. Luteinizing hormone (LH) and follicle stimulating hormone (FSH) are secreted by the anterior pituitary in response to gonadotropin releasing hormone released by the hypothalamus. LH stimulates Leydig cells to produce testosterone [Citation1,Citation5], which is need for the completion of meiosis and for spermiogenesis after the onset of puberty [Citation5]. Meanwhile, FSH helps initiate and maintain spermatogenesis by acting on Sertoli cells [Citation6].
Spermatogenesis has also been shown to be regulated by cell cycle proteins, including cyclin-dependent kinase inhibitors (CDKi) [Citation7–9]. The CDKi INK4 family includes p16Ink4a, p15Ink4b, p18Ink4c, and p19Ink4d, all of which primarily act by binding CDK4 and CDK6 to inhibit their activity, and thus arrest progression through the G1/S checkpoint of the cell cycle [Citation10,Citation11]. While mutations/deletions in most of the INK4 family result in an increase in the incidence of tumors [Citation10], an effect on spermatogenesis was also noted for Ink4c and Ink4d proteins. Specifically, Ink4c-/- mice exhibit failure of appropriate Leydig cells differentiation resulting in hyperplasia, low testosterone levels and high testicle weights, with elevated sperm counts but no obvious effect on fertility [Citation8]. On the other hand, Ink4d-/- male mice show testicular atrophy with increased apoptosis of germ cells, but also without affecting fertility [Citation9]. However, Ink4c-/- Ink4d-/- double knockout male mice are infertile, with extensive testicular atrophy and degeneration [Citation8]. This observed phenotype has been proposed to be a consequence of delayed differentiation of spermatogonia, which then undergo p53-independent apoptosis instead of entering meiosis [Citation8].
One of the most investigated tumor suppressor proteins, p53, also functions in cell cycle regulation, through its effects on transcription regulation in response to DNA damage and cell stress, resulting in DNA repair, cellular senescence, growth suppression, or apoptosis [Citation12,Citation13]. During normal spermatogenesis, p53 is expressed in the intermediate layer of the seminiferous tubules, in spermatocytes and round spermatids, suggesting that it might play a role in regulation of this intricate process [Citation14]. Indeed, p53-/- mice seem to have a higher percentage of abnormal sperm morphology when compared to wild-type counterparts, with an increased number of A1 spermatogonia suggesting an increase in early proliferation, but with no noticeable effect on male fertility [Citation15,Citation16].
In this report, we identify that p53-/- Ink4c-/- double knockout male mice have markedly reduced fertility, due to a defect in spermatogenesis. Our investigation reveals that absence of p53 partially reverses the Ink4c-/- phenotype of testicular and Leydig cell hyperplasia, and leads to persistent and prolonged delay in mitotic exit and meiotic progression, enhanced DNA damage and prolonged wave of apoptosis, culminating in abnormal and reduced sperm count. These findings shed light onto the cooperation between the p53 and the RB pathways in development and male fertility, and suggest that these players should be investigated in pathophysiology of human male infertility disorders.
Materials and methods
Mouse strains
Mouse strains that are p53 -/- (FVB.129-Trp53tm1Brn), Cdkn1a -/- (B6;126S2- Cdkn1atm1Tyj/J) (both from Jackson Laboratory, Maine), or p18Ink4c -/- [Citation7] were intercrossed, while maintained in a mixed C57BL/6 × 129Sv genetic background. p18Ink4c-/- females were bred with p53-null males to yield compound heterozygotes. Interbreeding generated wild-type and single- and double-null animals at the expected Mendelian frequencies. PCR for targeted alleles was used to verify mouse genotypes as previously described [Citation7,Citation17]. Animals were euthanized at defined time points for harvesting testes, in accordance with the American University of Beirut (AUB) Institutional Animal Care and Use Committee guidelines; all studies were approved by this committee.
Histological studies and Immunostaining
Testes were placed in 4% paraformaldehyde for at least 6 hours, then in 70% ethanol at 4°C and embedded in paraffin. 4 μm sections were deparaffinized, and stained with hematoxylin and eosin or processed for immunostaining. Antigen retrieval was performed in a steamer for 40 minutes in citrate antigen retrieval buffer (pH 6.0). Slides were incubated with anti-PCNA (Santa Cruz Biotechnology) or anti-phosphorylated H2AX (Millipore, California, CA) antibodies, followed by biotinylated secondary antibody; and detected using streptavidin conjugated to horseradish peroxidase and DAB substrate (DAKO, Carpinteria, CA). For immunofluorescence staining, anti-p450scc (Santa Cruz Biotechnology) or anti-phosphorylated H2AX (Millipore, California, CA) antibodies were detected with Cyanine 2 or Cyanine 3 secondary antibodies (Jackson Immunoresearch, West Grove, PA), and counterstained with DAPI (Vector Laboratories). Apoptotic cells were visualized by the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick 3′-end labeling (TUNEL) assay. TUNEL labeling was done using the Dead-End Cell Death Labeling Kit (Roche, Indianapolis, IN). The numbers of PCNA positive tubules and the total number of TUNEL positive cells per tubule were manually counted from 5 representative fields, at 40x magnification. For PCNA, a tubule was considered positive if it had more than 3 positive peripheral layers of cells. Image J software was used to quantify size of Leydig cells, by using the area quantitation tool and computing the mean area of Leydig cells per field, from 7 representative fields at 40x magnification for at least 3 mice of each genotype. Digital photomicrographs were obtained using a Zeiss 510 NLO multiphoton/ confocal laser scanning microscope. Composite images were constructed using Photoshop CS6 software (Adobe Systems, Mountain View, CA).
Assessment of sperm count, motility, and morphology
Three-month old male mice of the specified genotypes were used. For each mouse, two cauda epididymes were harvested at a similar time of day (between 12:00 p.m. and 2:00 p.m.). The sperm-containing fluid was squeezed out of the cauda, which was then cut into pieces. The sperm fluid and the pieces of cauda were suspended in 1 ml of Dulbecco's modified Eagle medium containing 25 mM HEPES buffer (pH 7.5) and 4 mg of bovine serum albumin per ml and were incubated at 37°C for 20 min. Suspensions of spermatozoa (20 μl) were fixed in 480 μl of 10% formalin. We used a hematocytometer to determine the number of spermatozoa.
For sperm motility assessment, the epididymides were isolated, cut and placed at 37°C in a cell incubator, in a tube containing 5 ml warm Dulbecco's Modified Eagles Medium (DMEM) containing 10% fetal bovine serum, 1% glutamine, and 1% Pen/Strep, and incubated for 30 minutes to allow spermatozoa to swim out. 100 μL of the solution were pipetted onto a glass slide and covered with a cover slip (size 22 × 22 mm), at room temperature. The motility of 100 spermatozoa was observed at a magnification of 40X and assessed by two different examiners, and graded as ‘progressive’, ‘nonprogressive’ or ‘immotile’, where progressive motility refers to sperm with linear or nonlinear forward motility (linear velocity ≥ 22 μm/s and velocity ≥ 5 μm/s), and nonprogressive motility to sluggish, circular or any pattern of movement with the absence of progression. Immotile sperm fail to move at all. The motility was expressed as the ratio of immotile to motile spermatozoa.
To assess the sperm morphology, 100 µL of the solution were spread onto a glass slide and allowed to air-dry at room temperature. The smears were then stained with Eosin Y stain. Two different examiners counted 100 cells per smear. A morphologically normal spermatozoon has an oval head and an acrosome covering 40%–70% of the head area. A normal spermatozoon has no neck, midpiece, tail abnormalities nor cytoplasmic droplets larger than 50% of the sperm head. The morphology was expressed as the ratio of abnormal to normal spermatozoa.
Results
Ink4c-/- p53-/- double knockout males, but not females, have markedly reduced fertility
We observed that, while Ink4c-/- and p53-/- male and female mice each are fertile with an average litter of 6 and 9 pups, respectively, and a pregnancy rate of 72% and 64% respectively, the double-knockout animals (Ink4c-/-p53-/-) had markedly reduced fertility, with a pregnancy rate of 8% and average litter size of 3 in the few successful pregnancies (, compare row 3 to rows 1–2). Pregnancy rates and litter sizes were comparable to controls when double-knockout females were mated to either single knockout males, or males retaining one copy of either p53 or Ink4c (, rows 4–7). However, pregnancy rates were extremely low when double knockout males were mated to either wild type or single knockout females (, rows 8–10), signifying male infertility as the underlying problem in double-knockout mating.
Table 1. Pregnancy rate and average litter size by mouse genotype.
Absence of p53 partially reverses the testicular hyperplasia seen in Ink4c-/- mice
To investigate the underlying mechanism of male infertility in this setting, we assessed testicular phenotype at post-natal day (P) 15, P19, P30, and P90, corresponding to Meiosis I, Meiosis II, first round of spermiogenesis, and adult spermatogenesis, respectively (see (A)).
Figure 1. p53 loss partially reverses the Ink4c-/- testicular phenotype. A) Spermatogenesis overview, with time points of the first wave of spermatogenesis in mice. The time line from birth onward indicates the temporal sequence of events. Red color denotes the timepoints at which phenotype analysis was performed in this study. Intervals in which mitotic cell division, meiosis I, meiosis II, and spermiogenesis occur are indicated above the time line, noting different stages during prophase of meiosis I. Spermatogonia are present within the seminiferous tubules at birth, and then give rise to spermatocytes, spermatids, and spermatozoa, at the indicated timepoints. B) Box plots representing the testis average weight (mg) for the WT (wild-type) (n = 7), p53-/- (n = 4), Ink4c-/- (n = 4) and Ink4c-/- p53-/- (n = 6) mice. Asterisks indicate significant p-value < 0.05. C) Representative staining for Hematoxylin and Eosin (H&E) in WT (wild-type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at 15 days postnatal (P15), 19 days postnatal (P19) and 3 months (m) of age. The lower row shows testes at a higher magnification (HM). Arrows denote the Leydig cells area. D) Representative staining for p450scc (middle), corresponding DAPI nuclear stain (left), and Merge (right), in WT (wild-type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at 3 months of age. E) Quantitation of the Leydic cells area compared to WT (wild type) in p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at 3 months of age. Asterisks indicate significant p-value < 0.05. NS denotes non-significant p-value.
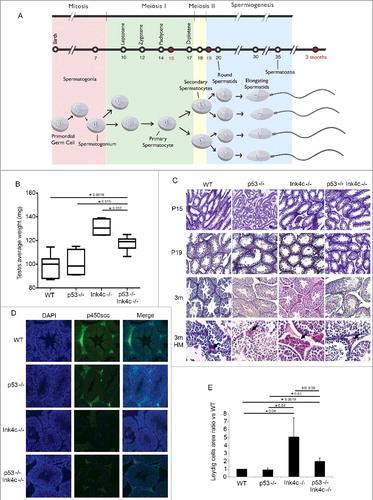
We compared testis size and weight across genotypes. While p53-/- mice were similar to wild-type (wt) mice in terms of testicular weight, Ink4c-/- mice showed an increased mean testis weight, as expected and previously described [Citation8] ((B)). Interestingly, loss of p53 partially reversed this phenotype, with mice doubly deficient for both Ink4c and p53 exhibiting an intermediate mean testis weight which is significantly higher than that of wt and p53-/- mice, but lower than that of Ink4c-/- mice ((B)).
Previous studies had demonstrated that testes of Ink4c-/- animals show Leydig cell hyperplasia [Citation8]. Histologic comparison of testes from wt, p53-/-, Ink4c-/-, and Ink4c-/-p53-/- mice at P15, P19 and P90 revealed that the hyperplasia of the Leydig cells seen in Ink4c-/- animals, which is most pronounced in adulthood, was partially reversed in the Ink4c-/-p53-/- double knockout animals ((C,E)).
The onset of mature Leydig cell function at puberty is characterized by their ability to terminally differentiate and produce testosterone and other androgens. To evaluate whether double knockout animals show a defect in Leydig cell differentiation, we assessed for P450scc expression, a steroidogenic enzyme responsible for conversion of cholesterol to pregnenolone, which is the first and rate-limiting step in the steroidogenic pathway [Citation18]. Similar to what has been previously reported [Citation8], we found reduced levels of P450ssc immunoreactivity in Leydig cells of Ink4c-/- mice ((D)). Interestingly, and as for testicular size, the phenotype of double-knockout animals was intermediate, with a decreased but not absent expression of P450scc ((D)), suggesting a partial reversal of the effects of Ink4c loss on Leydig cell differentiation as well as hyperplasia in absence of p53.
Ink4c-/- p53-/- double knockout mice have low sperm counts and a high proportion of immotile, sluggish, and abnormal sperm
Sperm count, mobility and morphology play a determining role in fertility [Citation19], therefore we anticipated that low sperm counts, reduced sperm mobility or morphological abnormalities might contribute to the observed infertility in the double knockout mice. It has been previously described that Ink4c-/- mice show a significantly increased sperm count as compared to wild-type mice, but maintain normal fertility [Citation8]. Consistently, we found that the number of spermatozoa in fluid extruded from the epididymides of Ink4c-/- males was significantly greater than that of both wild type and p53-/- males ((A)). Strikingly, p53-/-Ink4c-/- double knockout infertile males had much lower sperm count compared to the control genotypes ((A)). Importantly, only 3 tested double knockout male mice had sperm counts comparable to controls (see 3 outliers in (A)), and 2 of these 3 mice were included in the breeding study and were responsible for 2 of the 3 successful pregnancies observed and reported in .
Figure 2. Effect of dual p53 and Ink4c loss on sperm count, motility and morphology, and on ability of spermatozoa to exit mitosis. A) Box plots representing the sperm counts (106/ml) for WT (wild-type) (n = 5), p53-/- (n = 3), Ink4c-/- (n = 3) and Ink4c-/- p53-/- mice (n = 10, 3 outliers). Asterisks indicate significant p-value < 0.05. B) Box plots representing the ratio of immotile to motile sperm for WT (wild-type) (n = 5) and Ink4c-/- p53-/- mice (n = 6, 1 outlier). Asterisks indicate significant p-value < 0.05. C) Box plots representing the ratio of abnormal to normal sperm for WT (wild-type) (n = 5) and Ink4c-/- p53-/- mice (n = 6, 1 outlier). Asterisk indicates significantp-value < 0.05. D) Representative Eosin Y staining of WT (wild-type) and Ink4c-/- p53-/- sperms at 3 months of age. 1 denotes Free head, 2: Free tail, 3: Broken neck, 4: Dag defect, 5: Hook defect and 6: Tail-stump sperms. E) Representative Immunostaining for PCNA in WT (wild-type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at 15 days postnatal (P15), 19 days postnatal (P19) and 3 months (m) of age. The last row shows tubules at a higher magnification (HM). F) Box plots representing the percentage (%) of PCNA positive tubules compared to the total number of tubules per field in WT (wild-type) (n = 4), p53-/- (n = 4), Ink4c-/- (n = 4) and Ink4c-/- p53-/- mice(n = 7, 3 outliers). For quantification of difference, a tubule was considered positive if it had more than 3 positively stained peripheral layers of cells. Asterisks indicatesignificant p-value < 0.05. NS denotes non-significant p-value.
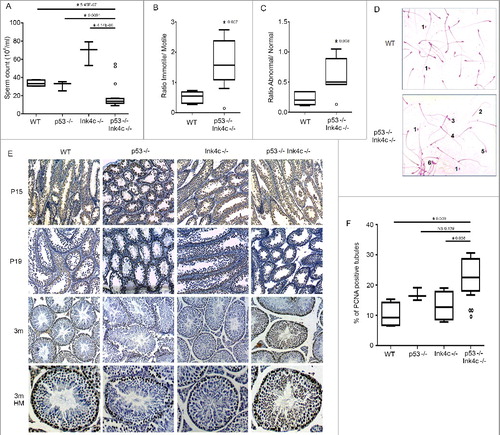
We then evaluated the motility of sperm of the different genotypes, defined as their ability to propel themselves forward. Motility is typically categorized into three grades: Normal sperm categorized as having progressive rapid motility swim fast in a straight line, or tend to travel forward in a curved motion. Nonprogressive motile sperm do not move forward despite the fact that they move their tails, and immotile sperm fail to move at all. For fertility, the percentage of normal progressively motile sperm should be over 32%, whereas reduced sperm motility beyond this level is termed asthenozoospermia [Citation19]. Indeed, we found that double knockout mice show a high ratio of immotile to motile spermatozoa as compared to wt animals ((B)). Interestingly, one of the 2 male mice that had successfully sired a pregnancy was included in this analysis, and showed an outlier value similar to that of the wild-type mice ((B), see outlier), further implicating these findings in the responsibility for reduced fertility.
We next evaluated sperm morphology, as morphological defects of sperm can also be important factors in infertility [Citation20]. Among morphological sperm abnormalities are head defects including round head sperms, multiple heads and tail, pinhead and broken neck, while abnormalities in the tail include nonflagellated free head, multiple tails, tail-stumps, and dag defects [Citation21]. Indeed, we found that the morphology of residual epididymal spermatozoa in the double-null mice was abnormal, having a greater ratio of abnormal to normal spermatozoa compared to wild-type males ((C,D)). Similar to what was observed for sperm count, the one male mouse that had successfully sired a pregnancy showed an outlier value similar to that of the wild-type mice ((C), see outlier).
Mice lacking both genes show a prolonged delay in spermatozoa entry into meiosis
Because Ink4c and p53 are cell cycle regulators [Citation11,Citation13], we anticipated that the loss of both inhibitors might affect premeiotic spermatogonial proliferation. To detect mitotically active spermatogonia, sections of testes were stained with PCNA, a marker of proliferation expressed in S phase [Citation22]. Up to P15, PCNA-positive cells were detected in testicular tubular cells of all genotypes ((E)). By P19, the number of PCNA-positive cells in tubules decreased in all genotype backgrounds, suggesting that many cells had exited the mitotic cycle. By adulthood, germ cell proliferation was restricted to a monolayer of cells along the basement membrane in the majority of seminiferous tubules in wild-type animals, but persisted in multiple cell layers in double knockout animals ((E), lower 2 panels). Quantitation of PCNA-positive tubules at the 3-month time-point showed that p53-/-Ink4c-/- mice had higher percentage of tubules with active proliferation, as compared to wild type and Ink4c-/- genotypes ((F)), whereas p53-/- males also had a higher percentage of positive tubules as compared to wild type (but not Ink4c-/-) males. This persistence of mitotic activity of the spermatogonial population in 3-month-old doubly deficient mice suggests that loss of both tumor suppressors leads to continuation of the mitosis phase in spermatogonia, and thus a delay in meiosis entry throughout adulthood.
Mice lacking both genes have an increase in spermatozoa apoptosis and evidence of DNA damage in spermatids
In testes from wild-type mice, as the first cohort of germ cells reaches the pachytene stage at P14 or P15, apoptosis of spermatogonia increases but then declines by P19 as some cells complete meiosis I [Citation4]. This wave of apoptosis is reflected by a quantifiable number of cells with condensed nuclei and DNA free ends detected by TUNEL assay ((A), left panel). We found that, while this wave of apoptosis is similar in p53-/- animals to that in wt mice, it was somewhat prolonged in Ink4c-/- mice ((A), compare second and third panels to first panel). However, in double-null mice, apoptosis persisted into adult mouse age ((A), right panel). Quantitation of the number of tubules containing TUNEL-positive cells showed that both Ink4c-/- single knockout and Ink4c-/-p53-/- double knockout testes show a prominence of apoptosis at P15 and P19, when compared to wt and p53-/- testes ((B)). However, only the double knockout testes show persistent apoptosis in testes of adult mice ((B)).
Figure 3. Mice lacking both genes show persistent spermatozoa apoptosis into adulthood as well as DNA damage in spermatids. A) Representative TUNEL staining (right) with its corresponding DAPI nuclear stain (left) in WT (wild type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at postnatal days P15, P19, and 3 months (m) of age. B) Quantitation of the mean number of apoptotic cells per tubule in WT (wild type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at the indicated ages. Asterisks indicate significant p-value < 0.05. NS denotes non-significant p-value. C) Representative Immunostaining for phospo-H2AX in WT (wild type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- testis at 3 months of age. Lower panel shows a higher magnification for representative images of WT (wild type) and Ink4c-/- p53-/- testes. D) Graph representing the percentage (%) of pH2AX positive tubules compared to the total number of tubules per field in WT (wild-type), p53-/-, Ink4c-/- and Ink4c-/- p53-/- mice at 3 months of age. Asterisks indicate significant p-value < 0.05.
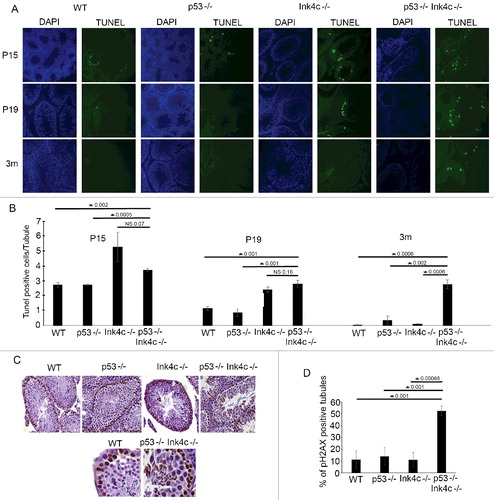
We considered whether the persistent proliferation noted in adult double knockout testes (see (D)) might be associated with DNA damage, as a possible cause for apoptosis in spermatogonia. Indeed, immunostaining for phospho-H2AX, a marker of DNA damage [Citation23], in testis at P90 showed positive staining in nuclei of spermatozoa of p53-/- Ink4c-/- double knockout mice, at all stages of maturation as well as in spermatids, a finding not seen in single knockout or wild type testes ((C)). Quantitation of pH2AX positive tubules showed that up to 50% of tubules stained for pH2AX in adult double knockout animals, compared to 10–15% of tubules in the wild type and single knockout animals ((D)).
The effects of p53 loss on spermatogonia are not due to loss of Cdkn1a
Another Cdk-inhibitor, Cdkn1a, is a well-studied downstream effector of p53 in cell cycle arrest [Citation24]. While Ink4c acts as an inhibitor of the cell cycle kinases Cdk4 and Cdk6, Cdkn1a acts primarily as an inhibitor of Cdk2 and Cdk1 [Citation25,Citation26]. We therefore investigated whether the effects of p53 on fertility in the setting of Ink4c loss are mediated through loss of induction of its downstream effector Cdkn1a. Previous studies have indeed noted some role for Cdkn1a in spermatogenesis control and cell cycle progression [Citation24,Citation27,Citation28]. However, we found that, in a total of 9 matings of Ink4c-/- Cdkn1a-/- mice, all 9 male mice sired offspring, with a pregnancy rate of 78% and an average litter size of 8 pups, thus comparable to wild type and single knockout mice (compare to ). We conclude that Cdkn1a is not responsible for the downstream effects of p53 in spermatogenesis control in this setting.
Discussion
Our work has uncovered a previously unrecognized interplay between p53 and Ink4c in spermatogenesis and male fertility. Consistent with their known roles as tumor suppressors, loss of p53, Ink4c, or both predisposes mice to different types of tumors [Citation10,Citation29–31]. However, their roles in spermatogenesis and fertility are less well defined. Male p53-/- mice are fertile with no evidence of defects in spermatogenesis [Citation15,Citation30], beyond a noted increase in number of early (A1) spermatogonia, suggesting an increase in proliferation at that stage [Citation15,Citation30]. On the other hand, Ink4c-/- mice have morphologic abnormalities in male testes, including testicular hypertrophy, defects in Leydig cell differentiation, and significantly increased sperm count; however they have normal fertility [Citation8]. Our findings now implicate p53 in maintaining fertility in the setting of Ink4c loss, as double knockout mice have partial reversal of the morphologic abnormalities seen in Ink4c-/- testes, including testes size and Leydig cell differentiation defect. However, the end result is a marked decrease in sperm count, and extensive abnormalities in sperm morphology and motility.
On a cellular level, we found that testes from double knockout mice show a prolongation of the period of spermatozoa proliferation and secondary delay of entry into meiosis, with accumulation of DNA damage in spermatozoa and spermatids, and persistent apoptosis continuing into adult male life. A wave of apoptosis is normally seen during the first wave of spermatogenesis. One function seems to be preventing cell crowding in seminiferous tubules and maintaining a proper ratio of differentiating spermatogonia to Sertoli cells [Citation32]. Another possible function is to act as a quality control mechanism, removing germ cells that could give rise to abnormal spermatozoa [Citation16]. While this wave of apoptosis seems to be transiently prolonged in Ink4c-/- mice, it persists into adulthood in Ink4c, p53 double knockout animals.
The transient increased apoptosis in Ink4c-/- testes has been suggested to be secondary to the observed transient delay in meiotic progression [Citation8]. Our results now suggest that p53 plays an important role in mitigating this effect, as loss of p53 results in further prolongation of meiosis entry, continued proliferation past P19, accumulation of cells with DNA damage, and persistent p53-independent apoptosis in adult testes, along with abnormal sperm cell morphology and motility, and male infertility.
The prolonged proliferation seen in Ink4c/p53 double knockout mice is not present in either single knockout genotype. This is different from the phenotype reported upon dual loss of another CDK4 inhibitor, Ink4a, and an upstream activator of p53, p19Arf. Similar to Ink4c, loss of Ink4a results in testicular hyperplasia, increase in sperm count, and increase in proliferation of early spermatogonia [Citation33]. However, the Leydig cell hyperplasia and differentiation defect seen in Ink4c-/- mice has not been reported in Ink4a-/- mice, suggesting an additional non-overlapping role for Ink4c in this setting. Loss of Arf leads to increased DNA damage in spermatogonia, which results in p53-mediated apoptosis, decreased testes size and reduced sperm count, but normal mouse fertility [Citation33]. Dual Ink4a/Arf knockout mice show increase in proliferation of spermatogonia similar to that observed in Ink4a-/- animals, and a persistent increase in pH2AX and induction of apoptosis similar to that seen in Arf-/- animals [Citation33]. In this manner, the Ink4a/Arf-/- double knockout results in normalization of testicular weight and sperm count, likely due to the independent but additive effects of both phenotypes. Notably, all 3 genotypes (Ink4a-/-, Arf-/- and double knockout) have normal fertility [Citation33]. In contrast, loss of p53 in Ink4c-/- animals leads to DNA damage, apoptosis, decreased (normalized) testes weight, decreased sperm counts, and markedly reduced fertility, which are not present in either single knockout phenotype. Therefore, unlike the case for Arf and Ink4a, which seem to act independently, p53 and Ink4c act in series in this setting, where p53 is necessary to prevent DNA damage and ensuing apoptosis in Ink4c-/- spermatogonia. The exact mechanism by which Ink4c loss leads to DNA damage accumulation (which is ameliorated by p53) is still unclear. Notably, both p53-dependent and p53-independent mechanisms of apoptosis have been described in testicular germ cells upon exposure to stress [Citation15,Citation34,Citation35].
Interestingly, absence of p53 also partially reverses the Leydig cell hyperplasia seen upon Ink4c loss, and allows normalization of Leydig cell differentiation defect. The previous study by Zindy et al [Citation8], and as our study replicates, shows that Ink4c is important in Leydig cell biology and differentiation, as well as in early meiosis exit in spermatogonia. Our study is the first to show a role for p53 in Leydig cell differentiation and hyperplasia in the setting of Ink4c loss, but the exact mechanistic pathway by which this occurs needs to be further elucidated. Also, since p53 loss affects both Leydig cells and spermatogonia in Ink4c-/- animals, the relative importance of the two cell types on the final infertility phenotype is not clear, although we suspect that the role of spermatogonia may be the most important, due to the findings of DNA damage, apoptosis, and abnormal sperm morphology.
The CDK2 inhibitor Cdkn1a is a well-characterized downstream effector of p53 in cell cycle control [Citation25,Citation26]. Previous studies have demonstrated a role for Cdkn1a in spermatogonial stem cell renewal and differentiation [Citation24]. Cdkn1a is expressed in quiescent primordial germ cells, spermatogonia, and supporting cells in the marbled newt, with a direct effect of p53 expression on levels of Cdkn1a and cell cycle arrest [Citation28]. In the mouse and the rat, Cdkn1a is expressed in spermatocytes and spermatids [Citation27,Citation36]. However, our results showed that it does not play a role in mediating effects of p53 loss on fertility in the Ink4c-/- setting. Indeed, p53 may act on the cell cycle independently of Cdkn1a, as previously shown in certain forms of senescence [Citation37,Citation38] at least partly through induction of Dec1 [Citation38], and cell cycle exit in response to DNA damage via upregulation of other downstream effectors such as TIGAR [Citation39], FOXM1 [Citation40], or direct repression of CDC20 [Citation41] and Cyclin B1 [Citation42]. Identifying the exact downstream effectors of p53 in the testis may provide further insights into the specific interplay among cell cycle proteins, DNA damage response, and differentiation pathways during spermatogenesis.
In male infertility disorders in humans, it is well recognized that there is a correlation between presence of markers of DNA damage, and abnormalities in sperm count, morphology, and motility [Citation43,Citation44], and therefore a causation effect may well be likely. However, no studies have yet characterized the underlying mechanisms, including the role of cell cycle pathways such as those converging on RB1 and p53, in disorders of male fertility. Our work suggests that cell cycle pathways such as p53 and Ink4c may be important candidates to investigate in such infertility conditions.
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Acknowledgments
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The Saab laboratory is partially funded by the American Lebanese Syrian Associated Charities (ALSAC) and International Outreach Program at St Jude Children's Research Hospital, Memphis, TN, through a development grant to the American University of Beirut, in Beirut, Lebanon.
Related Research Data
References
- Hess RA, Renato de Franca L. Spermatogenesis and cycle of the seminiferous epithelium. Adv Exp Med Biol. 2008;636:1–15. PMID:19856159
- Bellve AR, Cavicchia JC, Millette CF, et al. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J Cell Biol. 1977;74:68–85. doi:10.1083/jcb.74.1.68. PMID:874003
- Nebel BR, Hackett EM. Synaptinemal complexes in primary spermatocytes of the mouse: the effect of elevated temperature and some observations on the structure of these complexes in control material. Z Zellforsch Mikrosk Anat. 1961;55:556–565. doi:10.1007/BF00325417. PMID:14478699
- McCarrey JR. Development of the germ cell. Cell and Molecular Biology of the Testis. New York, NY: Oxford University Press; 1993. p. 58–89.
- O'Shaughnessy PJ. Hormonal control of germ cell development and spermatogenesis. Semin Cell Dev Biol. 2014;29:55–65. doi:10.1016/j.semcdb.2014.02.010. PMID:24598767
- Sairam MR, Krishnamurthy H. The role of follicle-stimulating hormone in spermatogenesis. Arch Med Res. 2001;32:601–608. doi:10.1016/S0188-4409(01)00328-9. PMID:11750736
- Latres E, Malumbres M, Sotillo R, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. doi:10.1093/emboj/19.13.3496. PMID:10880462
- Zindy F, den Besten W, Chen B, et al. Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18(Ink4c) and p19(Ink4d). Mol Cell Biol. 2001;21:3244–3255. doi:10.1128/MCB.21.9.3244-3255.2001. PMID:11287627
- Zindy F, van Deursen J, Grosveld G, et al. INK4d-deficient mice are fertile despite testicular atrophy. Mol Cell Biol. 2000;20:372–378. doi:10.1128/MCB.20.1.372-378.2000. PMID:10594039
- Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Bioch Bioph Acta. 2002;1602:73–87. PMID:11960696
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi:10.1101/gad.9.10.1149. PMID:7758941
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi:10.1038/nrc2723. PMID:19776744
- Lodish H, Berk A, Zipursky SL, et al. Mutations affecting genome stability. Molecular Cell Biology. New York: W. H. Freeman; 2000.
- Bornstein C, Brosh R, Molchadsky A, et al. SPATA18, a spermatogenesis-associated gene, is a novel transcriptional target of p53 and p63. Mol Cell Biol. 2011;31:1679–1689. doi:10.1128/MCB.01072-10. PMID:21300779
- Beumer TL, Roepers-Gajadien HL, Gademan IS, et al. The role of the tumor suppressor p53 in spermatogenesis. Cell Death Differ. 1998;5:669–677. doi:10.1038/sj.cdd.4400396. PMID:10200522
- Yin Y, Stahl BC, DeWolf WC, et al. p53-mediated germ cell quality control in spermatogenesis. Dev Biol. 1998;204:165–171. doi:10.1006/dbio.1998.9074. PMID:9851850
- Skapek SX, Lin SC, Jablonski MM, et al. Persistent expression of cyclin D1 disrupts normal photoreceptor differentiation and retina development. Oncogene. 2001;20:6742–6751. doi:10.1038/sj.onc.1204876. PMID:11709709
- Payne AH, Youngblood GL. Regulation of expression of steroidogenic enzymes in Leydig cells. Biol Reprod. 1995;52:217–225. doi:10.1095/biolreprod52.2.217. PMID:7711191
- WHO laboratory manual for the examination and processing of human semen. 5th ed. Geneva: World Health Organization; 2010. ISBN 978 92 4 154778 9
- Guzick DS, Overstreet JW, Factor-Litvak P, et al. Sperm morphology, motility, and concentration in fertile and infertile men. N Engl J Med. 2001;345:1388–1393. doi:10.1056/NEJMoa003005. PMID:11794171
- Chenoweth PJ. Genetic sperm defects. Theriogenology. 2005;64:457–468. doi:10.1016/j.theriogenology.2005.05.005. PMID:15993939
- Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: association with DNA replication sites. J Cell Biol. 1987;105:1549–1554. doi:10.1083/jcb.105.4.1549. PMID:2889739
- Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi:10.1038/nrc2523. PMID:19005492
- Kanatsu-Shinohara M, Takashima S, Shinohara T. Transmission distortion by loss of p21 or p27 cyclin-dependent kinase inhibitors following competitive spermatogonial transplantation. Proc Natl Acad Sci U S A. 2010;107:6210–6215. doi:10.1073/pnas.0914448107. PMID:20308578
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi:10.1038/nrc2657. PMID:19440234
- Lin J, Reichner C, Wu X, et al. Analysis of wild-type and mutant p21WAF-1 gene activities. Mol Cell Biol. 1996;16:1786–1793. doi:10.1128/MCB.16.4.1786. PMID:8657154
- Beumer TL, Roepers-Gajadien HL, Gademan LS, et al. P21(Cip1/WAF1) expression in the mouse testis before and after X irradiation. Mol Reprod Dev. 1997;47:240–247. doi:10.1002/(SICI)1098-2795(199707)47:3%3c240::AID-MRD2%3e3.0.CO;2-L. PMID:9170103
- Ricote M, Alfaro JM, Garcia-Tunon I, et al. Control of the annual testicular cycle of the marbled-newt by p53, p21, and Rb gene products. Mol Reprod Dev. 2002;63:202–209. doi:10.1002/mrd.10167. PMID:12203830
- Franklin DS, Godfrey VL, Lee H, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–2911. doi:10.1101/gad.12.18.2899. PMID:9744866
- Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi:10.1016/S0960-9822(00)00002-6. PMID:7922305
- Zindy F, Nilsson LM, Nguyen L, et al. Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res. 2003;63:5420–5427. PMID:14500377
- de Rooij DG, Grootegoed JA. Spermatogonial stem cells. Curr Opin Cell Biol. 1998;10:694–701. doi:10.1016/S0955-0674(98)80109-9. PMID:9914171
- Churchman ML, Roig I, Jasin M, et al. Expression of arf tumor suppressor in spermatogonia facilitates meiotic progression in male germ cells. PLoS Genet. 2011;7:e1002157. doi:10.1371/journal.pgen.1002157. PMID:21811412
- Perez GI, Knudson CM, Leykin L, et al. Apoptosis-associated signaling pathways are required for chemotherapy-mediated female germ cell destruction. Nat Med. 1997;3:1228–1232. doi:10.1038/nm1197-1228. PMID:9359697
- Yin Y, Stahl BC, DeWolf WC, et al. P53 and Fas are sequential mechanisms of testicular germ cell apoptosis. J Androl. 2002;23:64–70. doi:10.1002/jand.2002.23.1.64. PMID:11780924
- Hendry JH, West CM. Apoptosis and mitotic cell death: their relative contributions to normal-tissue and tumour radiation response. Int J Radiat Biol. 1997;71:709–719. doi:10.1080/095530097143716. PMID:9246185
- Castro ME, del Valle Guijarro M, Moneo V, et al. Cellular senescence induced by p53-ras cooperation is independent of p21waf1 in murine embryo fibroblasts. J Cell Biochem. 2004;92:514–524. doi:10.1002/jcb.20079. PMID:15156563
- Qian Y, Zhang J, Yan B, et al. DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J Biol Chem. 2008;283:2896–2905. doi:10.1074/jbc.M708624200. PMID:18025081
- Madan E, Gogna R, Kuppusamy P, et al. TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex. Br J Cancer. 2012;107:516–526. doi:10.1038/bjc.2012.260. PMID:22782351
- Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009;28:4295–4305. doi:10.1038/onc.2009.282. PMID:19749794
- Banerjee T, Nath S, Roychoudhury S. DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res. 2009;37:2688–2698. doi:10.1093/nar/gkp110. PMID:19273532
- Innocente SA, Lee JM. p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett. 2005;579:1001–1007. doi:10.1016/j.febslet.2004.12.073. PMID:15710382
- Schulte RT, Ohl DA, Sigman M, et al. Sperm DNA damage in male infertility: etiologies, assays, and outcomes. J Assist Reprod Genet. 2010;27:3–12. doi:10.1007/s10815-009-9359-x. PMID:20012685
- Zini A, Libman J. Sperm DNA damage: clinical significance in the era of assisted reproduction. CMAJ. 2006;175:495–500. doi:10.1503/cmaj.060218. PMID:16940270