
Toll-like receptor (TLR) signaling is critical to hematopoietic stem cell (HSC) function during normal differentiation and in response to host pathogens. During a normal immune response, transient TLR activation of immune effector cells mediates canonical NF-κB and MAPK signaling, eliciting expression of inflammatory-related genes as part of a highly coordinated temporal response. However, during TLR stimulation of HSC, myeloid-biased differentiation occurs and, if unresolved, results in HSC exhaustion, cytopenias, and bone marrow failure. The characteristic changes in bone marrow physiology stemming from prolonged TLR activation mirror the consequences of impaired HSC function associated with myelodysplastic syndromes (MDS), a clonal HSC malignancy defined by blood cytopenia, myeloid cell dysplasia, ineffective hematopoiesis, and a propensity for transformation to acute myeloid leukemia (AML). Thus, it is not surprising that chronic innate immune signaling downstream of TLR is implicated in the pathogenesis of MDS and other premalignant hematologic disorders.
Several molecular mechanisms and genetic alterations contribute to chronic innate immune signaling in MDS HSC, which converge on the central mediator of innate immune signaling, TRAF6. One notable example of TRAF6 dysregulation is in MDS with deletion of chromosome 5q (del(5q) MDS), which involves hemizygous deletion of miR-146a and TIFAB. Deletion of miR-146a increases TRAF6 mRNA and translation, while loss of TIFAB increases TRAF6 protein stability, thus resulting in overexpression and activation of TRAF6 in MDS HSC [Citation1,Citation2]. Collectively, TRAF6 is overexpressed in ∼40% of normal karyotype or del(5q) HSC from MDS patients as compared to HSC cells from healthy individuals [Citation3].
More recently, our studies have systematically explored the molecular and cellular consequences of TRAF6 overexpression on HSC [Citation3]. To model sustained TRAF6 overexpression and genetically-driven chronic TLR activation observed in MDS HSC, transgenic mice were generated that overexpress TRAF6 in hematopoietic cells at disease-relevant levels. TRAF6 overexpression resulted in HSC defects that are cell-intrinsic and associated with myeloid-biased differentiation. Although NF-κB and immune signaling is associated with normal TRAF6 function, gene expression and molecular analyses indicated that immune and inflammatory responses were not upregulated in TRAF6-overexpressing HSC, suggesting that the hematopoietic phenotype following TRAF6 overexpression was not associated with activation of canonical NF-κB signaling and/or inflammation.
TRAF6 is an E3 ligase that catalyzes the formation of lysine (K) 63-linked ubiquitin (Ub) chains on substrates. As such, we searched for ubiquitinated substrates of TRAF6 that may be implicated in the pathogenesis of MDS by performing a global ubiquitin screen and in vitro ubiquitin reconstitution assays. Several spliceosome auxiliary factors were identified as TRAF6 substrates, including hnRNPA1, hnRNPK, hnRNPM, hnRNPG, and hnRNPD. Moreover, TRAF6 overexpression in HSC resulted in RNA splicing changes of hematopoietic-requisite genes. Thus, it is possible that TRAF6 signaling downstream of TLR coordinates RNA processing via ubiquitination of multiple RNA splicing factors. Specifically, we found that the spliceosome-associated function of hnRNPA1, for example, was altered as a result of TRAF6-mediated K63 ubiquitination and led to aberrant RNA splicing of Arhgap1, a Rho family GTPase, and subsequent Cdc42 activation (). Activation of the small RhoGTPase Cdc42 is largely responsible for the hematopoietic phenotype exhibited by mice that express TRAF6 at nearly identically elevated levels as MDS HSCs (). Elevated Cdc42 activation has been causally linked to ineffective HSC function upon aging [Citation4], which is especially intriguing given that aged HSC are suspected to be afflicted by persistent innate immune signaling and inflammation [Citation5]. Interestingly, we found that among genes that were alternatively spliced as a result of altered hnRNPA1 function, NF-κB binding motifs were enriched in the promoters of those that exhibited exon exclusion, providing further evidence of the cooperative relationship shared by chronic innate immune signaling and alternative splicing (). Collectively, these findings highlight the significance of splicing factor mutations and RNA isoform usage in MDS and AML and underscore the importance of RNA processing factor regulation through alternative splicing and/or posttranslational modification.
Figure 1. Overview of isoform usage in response to transient (left) or chronic TLR signaling (right). During a normal immune response, TRAF6 transiently catalyzes K63-linked ubiquitin (Ub) chains on RNA binding proteins, like hnRNPA1, which leads to differential splicing of target genes, such as Arhgap, and an appropriate balance of isoform expression. Chronic TLR activation results in TRAF6 overexpression and continuous ubiquitination of hnRNPA1 (1), which leads to skewed isoform usage of Arhgap (2) and subsequent alteration of downstream signaling pathways (3), such as the one involving the small G-protein GTPase Cdc42. Overactive Cdc42 signaling contributes to HSPC defects and myelodysplasia associated with bone marrow failure. Transcription of alternatively spliced genes was not exclusively driven by NF-κB and likely involves other transcription factors (TF). NF-κB signaling was not hyperactive following TRAF6 overexpression (indicated in grey).
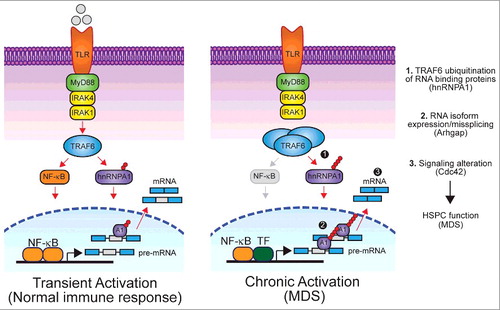
Posttranslational modifications are important for contributing to the fluidity of protein-protein and RNA-protein interactions to regulate mRNA processing and RNA splicing dynamics. While ubiquitin modification of splicing factors has been shown to control spliceosome assembly in yeast [Citation6], ubiquitination of hnRNPA1 by TRAF6, to the best of our knowledge, represents the first evidence of ubiquitination of splicing factors in mammalian cells. We propose a model in which TRAF6 couples NF-κB-induced transcription with pre-mRNA processing. That is, TLR stimulation not only results in TRAF6-mediated activation of NF-κB target genes, but also simultaneously mediates ubiquitination of RNA binding proteins, such as hRNPA1, resulting in selective changes in RNA isoform expression during the inflammatory response (). These findings align with recent observations about RNA processing during immune responses and normal differentiation. For example, T cell receptor (TCR) signaling in human and murine CD4+ T cells leads to hnRNPU-induced isoform switching of MALT1B to MALT1A, which in turn results in enhanced NF-κB signaling and allows for productive T cell activation [Citation7]. Interestingly, hnRNPA1 was also responsible for isoform switching of MALT1B to MALT1A. In addition, RNA processing is essential during normal granulocyte differentiation. During granulopoiesis, downregulation of the splicing component Snrpa correlates with increased intron retention (IR) and subsequent nonsense-mediated decay of many transcripts, including Lmnb1 [Citation8]. IR-induced downregulation of Lmnb1 transcript and protein levels results in reduced proliferation and allows for eventual differentiation into mature granulocytes.
Taken together, there are clear implications for developing our understanding of the broader impacts of RNA processing as it relates to disease pathogenesis. Specifically, the identification of recurrent somatic mutations of spliceosome genes in MDS and other hematologic malignancies suggests the significance of RNA splicing for disease phenotypes. In addition, our findings and those of other recent studies point to the possibility that altered RNA processing extends beyond patients with spliceosome gene mutations. Extant research, therefore, leads to the following hypothesis that should inform an underexplored avenue of research: that posttranslational modification of splicing regulators and alternative splicing represents a fine-tuned and combinatorial control mechanism for cellular responses and dysregulation of this system serves as a likely culprit in the pathogenesis of cancer.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors
Additional information
Funding
References
- Varney ME, Niederkorn M, Konno H, et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor–TRAF6 signaling. J Exp Med. 2015;212(11):1967–1985. doi:10.1084/jem.20141898. PMID:26458771.
- Varney ME, Choi K, Bolanos L, et al. Epistasis between TIFAB and miR-146a: neighboring genes in del(5q) myelodysplastic syndrome. Leukemia. 2017;31(7):1659. doi:10.1038/leu.2017.95. PMID:28386122.
- Fang J, Bolanos LC, Choi K, et al. Corrigendum: Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol. 2017;18(4):474. doi:10.1038/ni0417-474a PMID:28323261
- Bellare P, Small EC, Huang X, et al. A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol. 2008;15(5):444–451. doi:10.1038/nsmb.1401. PMID:18425143.
- Florian MC, Dörr K, Niebel A, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10(5):520–530. doi:10.1016/j.stem.2012.04.007. PMID:22560076.
- Esplin BL, Shimazu T, Welner RS, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. 2011;186(1):5367–5375. doi:10.4049/jimmunol.1003438. PMID:21441445.
- Meininger I, Griesbach RA, Hu D, et al. Alternative splicing of MALT1 controls signalling and activation of CD4+ T cells. Nat Commun. 2016;7:11292. doi:10.1038/ncomms11292. PMID:27068814.
- Wong JJ, Ritchie W, Ebner OA, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154(3):583–595. doi:10.1016/j.cell.2013.06.052. PMID:23911323.