
ABSTRACT
RCC1 associates to chromatin dynamically within mitosis and catalyzes Ran-GTP production. Exogenous RCC1 disrupts kinetochore structure in Xenopus egg extracts (XEEs), but the molecular basis of this disruption remains unknown. We have investigated this question, utilizing replicated chromosomes that possess paired sister kinetochores. We find that exogenous RCC1 evicts a specific subset of inner KT proteins including Shugoshin-1 (Sgo1) and the chromosome passenger complex (CPC). We generated RCC1 mutants that separate its enzymatic activity and chromatin binding. Strikingly, Sgo1 and CPC eviction depended only on RCC1's chromatin affinity but not its capacity to produce Ran-GTP. RCC1 similarly released Sgo1 and CPC from synthetic kinetochores assembled on CENP-A nucleosome arrays. Together, our findings indicate RCC1 regulates kinetochores at the metaphase-anaphase transition through Ran-GTP-independent displacement of Sgo1 and CPC.
Introduction
Centromeres are chromatin domains that play four critical roles during mitosis [Citation1]. First, centromeres are platforms for the assembly of kinetochores (KTs) that attach sister chromatids to the mitotic spindle. Second, Shugoshin (Sgo) proteins within the inner centromeric region (ICR) maintains the cohesion between the sister chromatid in mitosis [Citation2]. This persistent cohesion is essential for correct MT attachment and anaphase segregation of sister chromatids. Third, sister KT attachment to microtubules (MTs) from opposite poles generates tension across the ICR that modulates the spindle assembly checkpoint (SAC), a regulatory pathway that detects unattached KTs and prevents anaphase if they are present. A key SAC component is the chromosomal passenger complex (CPC), comprised of the Aurora B kinase (AurB), INCENP, Borealin, and Survivin [Citation3]. Notably, Shugoshin proteins and the CPC show interdependent localization [Citation4]. Finally, ICRs must be rapidly and synchronously re-modeled at anaphase onset, both to release sister chromatid cohesion and to redistribute the CPC to MTs at the spindle midzone [Citation3].
RCC1 is the guanine nucleotide exchange factor (GEF) for the Ran GTPase, which regulates multiple cellular processes [Citation5]. RCC1 binds chromatin throughout the cell cycle, and generates spatially defined patterns of Ran-GTP distribution. The interaction of RCC1 with chromatin changes dramatically at anaphase onset [Citation6,Citation7]. The RanBP1 protein controls RCC1 partitioning between its chromatin-bound and soluble pools in mitotic Xenopus egg extracts (XEEs) by forming a heterotrimeric complex with Ran and RCC1 (RRR complex) [Citation8]. Phosphorylation on RanBP1 at anaphase onset disassembles RRR complexes, increasing the amount of RCC1 on chromatin. Mimicking the anaphase wave of chromatin-bound RCC1 through addition of exogenous RCC1 to metaphase-arrested XEEs (CSF-XEEs) disrupts both KT composition and SAC activity in reactions containing unreplicated demembraned sperm nuclei (DSN) [Citation6].
To better understand how RCC1-chromatin association may be important at anaphase onset, we investigated ICR re-structuring, utilizing replicated chromosomes that possess physiologically paired sister KTs [Citation9]. We observed highly specific release of Sgo1 and CPC by RCC1, with minimal changes in outer KT components. To understand how RCC1 might control Sgo1 and CPC, we examined RCC1 mutants that were impaired either for enzymatic RanGEF activity or for binding to chromatin. Strikingly, enzymatically inactive RCC1 could evict Sgo1 and CPC as efficiently as wild type RCC1, while chromatin-binding mutants had no similar activity. Moreover, RCC1 was capable of evicting Sgo1 and CPC from CENP-A nucleosome array-coated beads [Citation10] in a manner that did not require its enzymatic activity. Together, our findings indicate RCC1 regulates KT composition at the metaphase-anaphase transition in a Ran-GTP-independent fashion through displacement of Sgo1 and CPC.
Results
Exogenous RCC1 evicts Sgo1 and CPC from KTs in mitotic XEEs
XEEs can be prepared under conditions that preserve the M-phase arrest of the intact egg by a meiosis-specific factor, called cytostatic factor (CSF). In the presence of nocodazole, unattached KTs of DSN generate SAC signals that maintain M-phase arrest even after the addition of CaCl2 to destruct CSF [Citation11]. DSN directly added to CSF-XEE has not undergone replication and thus cannot form paired sister KTs. RCC1 disrupts KTs of unreplicated DSN chromosomes, causing the release of a variety of outer KT components and inactivating the SAC [Citation6]. To examine the capacity of exogenous RCC1 to remodel replicated KTs, we added DSN to XEE that had been driven into interphase, allowing a complete round of DNA replication. Subsequent addition of a fresh aliquot of CSF-XEE returned the reaction to M-phase. We simultaneously added nocodazole to trigger SAC activation and examined the behavior of KT proteins by immunoblotting (IB) and immunofluorescence (IF) ((A)). We found that RCC1 efficiently evicted Sgo1 and AurB from the replicated chromosomes even when RCC1 was added at only three-fold exogenous concentrations ((B–D)), a level comparable to physiological RCC1 fluctuation on chromatin at anaphase onset in unperturbed cycling XEEs ((B), lower panel). Notably, outer KT components, including Bub1 and Mad2, were much less sensitive to increased RCC1 levels ((B–D)).
Figure 1. Exogenous RCC1 evicts Sgo1 and AurB from KTs in mitotic XEEs. (A) Experimental timeline. XEEs containing DSN (10,000 units/µl) were induced for DNA replication before driven back to mitosis. XEEs aliquots were analyzed by immunoblotting (IB) and immunofluorescence (IF) after indicated treatments. (B) Input reactions and chromatin fraction from untreated XEEs or XEEs with increasing concentrations of RCC1 (2, 6, or 20 μg/ml) were analyzed by IB analysis. Cycling XEEs were prepared and chromatin was isolated from different time points. Proteins content from both total extracts and chromatin fractions were analyzed by IB. The chromatin bound RCC1 intensity readings were quantified, normalized to first lane, and plotted in bottom panel. (C)(D) Chromatin from samples as in (B) were purified, stained with Hoechst 33342, and subjected to IF analysis against Bub1 (top, red) and (C) Sgo1 or (D) AurB (middle, green). Scale bar, 20 μm.
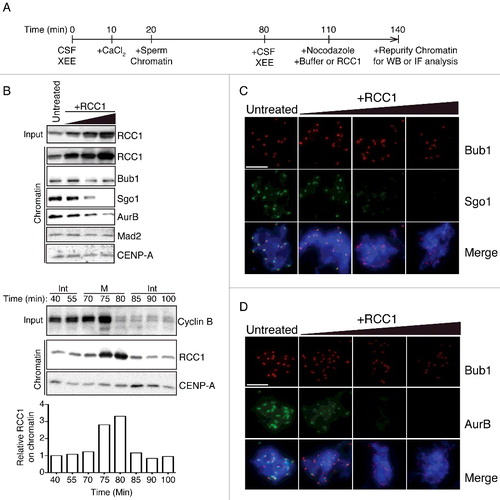
These data indicate that RCC1-mediated re-organization of replicated KTs is remarkably specific, preferentially evicting CPC and Sgo1 (). This finding contrasts with prior observations with unreplicated DSN [Citation6], where exogenous RCC1 caused a gross eviction of kinetochore proteins. To test the differences, we made systematic comparisons between unreplicated chromatin from CSF XEEs and replicated chromatin from cycled XEEs (Fig. S1). We found that Sgo1 and AurB at ICR were sensitive to exogenous RCC1 in both CSF and cycled XEEs. However, other outer kinetochore proteins including Bub1 and Mad2 were significantly affected only in CSF XEEs but not cycled XEEs (Fig. S1, left and right panels, lane 1&2). While unreplicated DSN efficiently form spindles in CSF-XEE, the geometry of their KTs is non-physiological. KTs in CSF XEEs were systematically compared with replicated KTs in cycled XEEs to reveal significantly different properties in a previous study [Citation12]. We believe that the earlier observations regarding KT remodeling by RCC1 primarily reflect RCC1's effect on Sgo1 and CPC, and that different responses of outer KT proteins to exogenous RCC1 can be explained for the distinct configurations of unreplicated versus replicated KTs.
Sgo1 and CPC recruitments were complete before RCC1 addition
The addition of exogenous RCC1 caused reductions of centromeric Sgo1 and AurB in cycled XEEs. However, it is possible that RCC1 was added before complete recruitment of those proteins to ICR. To exclude this possibility, we aliquoted cycled XEEs before RCC1 addition and checked the level of chromatin-associated Sgo1 and AurB ((A)). We found that at time T0, both Sgo1 and AurB have already been recruited to ICR ((B), lane 1; (C), column 1). Their levels at T30 were not changed by further incubation in control XEEs, but reduced only in XEEs with RCC1 addition ((B–C)).
Figure 2. RCC1 evicts Sgo1 and AurB without blocking their recruitment. (A) Experimental timeline. XEEs aliquots were taken before (T0) and after (T30) RCC1 addition (20 μg/ml) and were analyzed by IB and IF analysis. (B) Chromatin fraction from XEEs were isolated at T0 and T30 and analyzed by IB analysis. (C)(D) Chromatin from samples as in (B) were purified, stained with Hoechst 33342, and subjected to IF analysis against Bub1 (top, red) and (C) Sgo1 or AurB (D) Histone H2ApThr121 (middle, green). In parallel, CSF XEEs depleted of Bub1 were also cycled to mitosis and subjected to the same IF analysis. Scale bar, 20 μm.
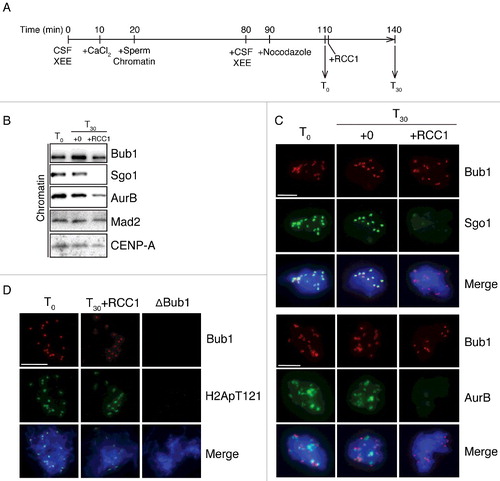
Sgo1 recruitment to ICR requires phosphorylation of Histone H2A at Thr121 by Bub1 kinase [Citation13]. An alternative explanation to Sgo1 level reduction is that the Thr121 might be inaccessible to Bub1 upon RCC1 addition. We checked phosphorylation status of this residue before and after adding exogenous RCC1 and found identical phosphorylation levels. Only immunodepletion of Bub1, but not RCC1 addition, could abolish the phosphorylation on Histone H2A Thr121 ((D)).
Together, we concluded that Sgo1 and AurB had been fully recruited to ICR before RCC1 addition. The exogenous RCC1 most likely evicted those proteins from ICR instead of blocking their recruitments.
Sgo1 and CPC eviction by RCC1 is independent of RanGEF activity
All previously identified functions of RCC1 depend on its capacity to generate Ran-GTP [Citation5]. We reasoned that if RCC1 evicted Sgo1 and CPC from KTs through Ran-GTP, we should be able to recapitulate this effect through addition of RanQ69L, a constitutively active Ran mutant [Citation14]. We added RanQ69L (0.2 mg/ml) to reactions containing replicated chromosomes in CSF-XEEs. After 30 min, we isolated chromosomes from reactions with and without RanQ69L and found no differences in either the levels or the localization of chromosome-associated Sgo1 and AurB ((A), lane 1 & 2; (B), column 1 & 2). Indeed, RanQ69L could not evict Sgo1 or AurB even when added to a concentration of 20 μM (Fig. S1, lane 5). These data indicated that Ran-GTP does not promote Sgo1 and CPC eviction.
Figure 3. Eviction of inner KT proteins by RCC1 is independent of RanGEF activity. (A) Buffer, RanQ69L (0.2 mg/ml), RCC1WT (40 μg/ml), or RCC1Ran (40 μg/ml) were added to mitotic XEEs containing chromatin (10,000 DSN/μl). Input reactions and chromatin fractions from each sample were examined by IB analysis. (B) Chromatin from samples as in (A) were purified, stained with Hoechst 33342, and subjected to IF analysis against Bub1 (row 2, red) and Sgo1 (row 3, green). Scale bar, 20 µm. (C) Either buffer or nocodazole (20 μg/ml) were added to CSF-XEEs, as indicated. DSN were added to a concentration of 10,000 units/μl. Buffer, RanQ69L (20 μM), RCC1WT (40 μg/ml), or RCC1Ran (40 μg/ml) were added as indicated. CaCl2 was added to each sample to a concentration of 0.1 mg/ml. XEE aliquots before and after CaCl2 treatment were examined by IB.
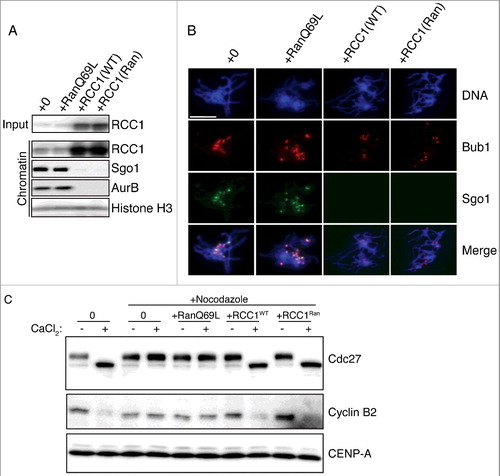
To confirm that the eviction of Sgo1 and CPC by RCC1 was independent of RCC1's enzymatic activity, we tested a RCC1 mutant (RCC1Ran) with no Ran binding affinity [Citation8]. We added identical amount of wild type RCC1 (RCC1WT) or RCC1Ran to cycled XEEs containing replicated chromosomes, and examined Sgo1 and CPC eviction as before. Strikingly, RCC1Ran evicted of both Sgo1 and AurB as efficiently as RCC1WT, as assayed on both by IB and IF of the isolated chromatin fraction ((A), lane 3 & 4; (B), column 3 & 4). We also tested RCC1Ran in combined with 20 μM RanQ69L. As expected, RanQ69L could not affect the protein eviction function of RCC1Ran (Fig. S1, lane 6). Together, these findings argue that RCC1's capacity to evict Sgo1 and CPC from mitotic chromosomes is independent of its role as a RanGEF.
Exogenous RCC1 disrupts the SAC in nocodazole-treated CSF-XEEs containing unreplicated DSN [Citation6]. We reasoned that if such SAC disruption were the result of Sgo1 and CPC eviction, it should be likewise insensitive to Ran-GTP levels. To test this idea, we added RanQ69L, RCC1WT or RCC1Ran to nocodazole-treated CSF-XEEs with unreplicated DNS, and took aliquots from each sample before and 30 min after CaCl2 addition. The mitotic status of these samples was determined by IB with antibodies against Cdc27 and Cyclin B2 ((C)). As expected, the control reaction without nocodazole exited from mitosis, showing Cdc27 dephosphorylation and Cyclin B degradation, while both markers remained static in the reaction containing nocodazole. RanQ69L did not cause M-phase exit of nocodazole-treated samples, indicating that elevated Ran-GTP levels did not provoke SAC override. By contrast, RCC1Ran caused Cdc27 dephosphorylation and Cyclin B2 degradation as efficiently as RCC1WT, indicating that RanGEF activity is not necessary for SAC disruption, consistent with the idea that earlier observations of SAC disruption reflect Sgo1 and CPC mis-localization.
RCC1 chromatin binding mutant with normal RanGEF cannot evict Sgo1 or AurB
Our data suggested that RCC1's regulation of Sgo1 and CPC is surprisingly independent of Ran-GTP production, which seemed to contradict with the inability of RCC1D182A mutant to evict centromeric proteins reported previoiusly [Citation6]. RCC1D182A mutant was known as a RanGEF inactive mutant [Citation15]. Here, we examined the chromatin affinity of RCC1D182A in XEEs. We added either wild type human RCC1 or RCC1D182A to M-phase XEEs and checked the bound RCC1 by immunofluorescence. We found that while hRCC1WT addition showed increased staining of RCC1 on chromatin, RCC1D182A could not be detected on chromatin by monoclonal antibodies against hRCC1 (3D11) (Fig. S2A). Thus, RCC1D182A is deficient in both RanGEF activity and chromatin affinity, undermining its suitability to determine which aspect is important in regulating centromeric composition. Therefore, we moved to make new RanGEF inactive RCC1 mutants that possess normal affinity to chromatin.
We generated Xenopus RCC1 mutants based on previous structural and functional studies of RCC1 [Citation16–18]. We generated a DNA binding mutant (RCC1DNA) and a Histone binding mutant (RCC1Hist) ((A)), and tested the nucleotide release activity of both mutants by incubating them with Ran that was loaded with [α-32P]GTP (Ran-[α-32P]GTP) and free, unlabeled GTP. After 30 min at room temperature, the remaining Ran-[α-32P]GTP was measured using a membrane retention assay ((B)). RCC1DNA or RCC1Hist (red and green lines) showed activities similar to RCC1WT (blue line), confirming that mutations in the chromatin-binding domains on RCC1 did not affect its enzymatic RanGEF function. By contrast, we observed almost no [α-32P]GTP release in reactions containing the RCC1Ran mutant (purple line).
Figure 4. Mutant analysis shows that RCC1 must bind chromatin for eviction of inner KT proteins. (A) Table showing names, mutations, chromatin affinity (measured in XEEs), and RanGEF activity (measured through in vitro assays) of wild type or mutant RCC1. (B) 1 µM recombinant Ran charged with [α-32P]GTP was incubated with 0, 0.1, 0.3, or 1 nM RCC1 at RT for 30 min, and exchange was monitored using a filter retention assay. Radioactivity retained on filters was measured by scintillation counter and plotted against RCC1 concentration as mean ± SEM (n = 3). (C) Analysis of chromatin binding for RCC1 mutants. RCC1 was immunodepleted from XEEs. DSN were added to a concentration of 10,000 units/μl. Wild type or mutated RCC1 was added back to depleted XEEs at concentrations of 6, 20, 60, 200, or 600 μg/ml. Chromatin was re-purified and subjected to quantitative IB with antibodies against RCC1. (D) Fluorescent intensity of RCC1 in chromatin fractions from samples as in (C) were acquired by Odyssey infrared imaging system and plotted against input RCC1 concentration as mean ± SEM (n = 3). (E) Buffer, RCC1WT (40 µg/ml), RCC1Hist (40 µg/ml) or RCC1DNA (40 µg/ml) were added to mitotic XEEs containing chromatin (10,000 DSN/µl). Input reactions and chromatin fractions from each sample were examined by IB analysis.
![Figure 4. Mutant analysis shows that RCC1 must bind chromatin for eviction of inner KT proteins. (A) Table showing names, mutations, chromatin affinity (measured in XEEs), and RanGEF activity (measured through in vitro assays) of wild type or mutant RCC1. (B) 1 µM recombinant Ran charged with [α-32P]GTP was incubated with 0, 0.1, 0.3, or 1 nM RCC1 at RT for 30 min, and exchange was monitored using a filter retention assay. Radioactivity retained on filters was measured by scintillation counter and plotted against RCC1 concentration as mean ± SEM (n = 3). (C) Analysis of chromatin binding for RCC1 mutants. RCC1 was immunodepleted from XEEs. DSN were added to a concentration of 10,000 units/μl. Wild type or mutated RCC1 was added back to depleted XEEs at concentrations of 6, 20, 60, 200, or 600 μg/ml. Chromatin was re-purified and subjected to quantitative IB with antibodies against RCC1. (D) Fluorescent intensity of RCC1 in chromatin fractions from samples as in (C) were acquired by Odyssey infrared imaging system and plotted against input RCC1 concentration as mean ± SEM (n = 3). (E) Buffer, RCC1WT (40 µg/ml), RCC1Hist (40 µg/ml) or RCC1DNA (40 µg/ml) were added to mitotic XEEs containing chromatin (10,000 DSN/µl). Input reactions and chromatin fractions from each sample were examined by IB analysis.](/cms/asset/c79888d9-d350-4eca-a37b-0bc218864fee/kccy_a_1442630_f0004_oc.jpg)
We tested the chromatin binding capacity of the RCC1 mutants by immunodepleting endogenous RCC1 from CSF-XEE and adding back various concentrations of each protein in the presence of 10,000 DSN/µl. We subsequently isolated the chromatin fraction from each reaction and measured the RCC1 level in individual sample by quantitative Western blotting ((C)). RCC1WT bound chromatin efficiently and saturated chromatin binding when added at a level roughly 30-fold higher than endogenous RCC1 concentrations ((D), blue line). RCC1Ran bound chromatin with an even higher efficiency than RCC1WT ((D), purple line), likely because RanBP1 sequesters a fraction of RCC1WT in RRR complexes that RCC1Ran does not form [Citation8]. Both RCC1DNA and RCC1Hist bound chromatin less efficiently than RCC1WT: the affinity of RCC1WT for chromatin was around 4-fold higher than RCC1DNA and more than 15-fold higher than RCC1Hist ((D), red and green lines). In parallel, we examined the interaction of each mutant with isolated nucleosome core particles by gel filtration chromatography. While RCC1WT and RCC1Ran co-fractionated with core particles, RCC1DNA and RCC1Hist did not, confirming their reduced affinity to nucleosomes (Fig. S2B). Collectively, these data show that RCC1DNA and RCC1Hist have reduced affinity for chromatin but normal RanGEF activity, while RCC1Ran has a normal affinity for chromatin but lacks RanGEF activity. Thus, we successfully separated RCC1's activity as a RanGEF from its activity as a chromatin-binding protein.
We then tested whether chromatin binding was important for RCC1 eviction of Sgo1 and CPC, we added RCC1WT, RCC1DNA or RCC1Hist to XEEs containing replicated DSN chromosomes. After incubation, we isolated the chromatin fraction and examined Sgo1 and AurB by Western blotting (E). While exogenous RCC1WT efficiently evicted both Sgo1 and AurB (lane 2), RCC1DNA or RCC1Hist were far less efficient at the same concentrations of added protein (lane 3 & 4). We also tested the RCC1 chromatin binding mutant's effect on outer kinetochore proteins. As expected, the addition of exogenous RCC1Hist did not change chromatin associated Bub1 and Mad2 (Fig. S1, lane 4). Together, our data argues that chromatin binding of RCC1 is indeed important for the removal of Sgo1 and AurB.
RCC1 evicts Sgo1 and CPC from KTs assembled on CENP-A-nucleosome arrays
To test whether RCC1 might evict Sgo1 and CPC by physical competition, we tested RCC1's capacity to remodel artificial KTs associated with CENP-A-containing nucleosome arrays. We assembled CENP-A nucleosome arrays and immobilized them on beads by strictly following the established protocols [Citation10] (Fig. S3A). We then added the beads into CSF-arrested XEEs and triggered the XEEs to enter mitosis ((A)). We confirmed the specificity of KT proteins recruitment by isolating Histone H3 nucleosome beads and hCENP-A nucleosome beads from cycled XEEs and analyzing the associated proteins by Western blotting. As expected [Citation10], we detected KT proteins on hCENP-A nucleosome beads but not on Histone H3 nucleosome beads. RCC1 was recruited to both types of beads (Fig. S3B). We also found xCENP-A was only detected on beads from cycled XEEs but not CSF XEEs, suggesting replication and new histone loading during cycling. Moreover, kinetochore proteins are only significantly recruited to beads in cycled XEEs but not in CSF XEEs (Fig. S3C).
Figure 5. RCC1 evicts proteins from KTs assembled on CENP-A nucleosome arrays. (A) Schematic diagram with timeline. Streptavidin beads conjugated with CENP-A-containing nucleosome array were added into CSF-XEEs and cycled to mitosis. The beads were either maintained in M-phase XEEs (B) or isolated in buffer (C). Wild type or mutated RCC1 proteins were added, and the beads were isolated. (B) Buffer, RCC1WT (40 µg/ml), RCC1Ran (40 µg/ml) or RCC1Hist (40 µg/ml) were added to 100 µl mitotic XEEs containing 10 µl CENP-A-containing nucleosome array beads, and incubated at RT for 30 min. Beads were isolated, eluted, and examined by IB. (C) 10 µl CENP-A-containing nucleosome array beads were isolated from mitotic XEE and resuspended in 100 µl ice-cold washing buffer. Buffer, RCC1WT (40 µg/ml), RCC1Ran (40 µg/ml), or RCC1Hist (40 µg/ml) were added and incubated on ice for 5 min. Beads were isolated again, eluted, and examined by IB. (D) Model for the role of RCC1 in Sgo1 and CPC eviction: Prometaphase/metaphase: RCC1 is partitioned between an active, chromatin-bound pool (green) and an inactive pool (gray). The inactive pool is associated with RRR complexes that also contain RanBP1 and nucleotide-free Ran (green). Anaphase: phosphorylation of RanBP1 (asterisk) releases RCC1 from the RRR complex. The free RCC1 is recruited to chromatin and evicts inner KT proteins, including Sgo1, by physical competition.
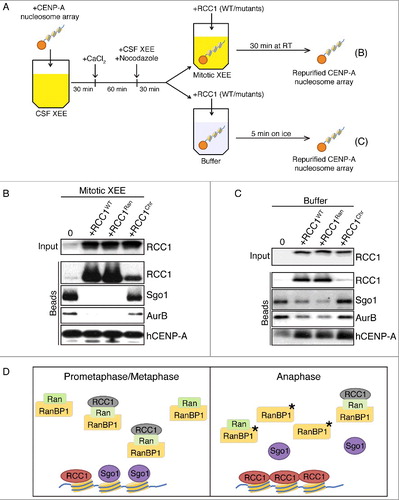
To test whether exogenous RCC1 could evict KT proteins assembled onto hCENP-A nucleosome beads, we added either wild type or mutated RCC1 proteins into the reaction. After 30 min incubation, proteins on beads were isolated and analyzed by IB ((A), top). Sgo1 and AurB were removed from hCENP-A nucleosome beads by the addition of either RCC1WT or RCC1Ran ((B)). However, RCC1Chr, combining mutants on Histone binding and DNA binding (R223A/K238A/K240A/R244A), was unable to evict Sgo1 and AurB, recapitulating that same pattern of eviction by RCC1 that we observed with DSN.
Lastly, we assayed whether RCC1 could evict KT proteins after the beads were isolated from XEEs. We assembled KT proteins to hCENP-A nucleosome beads in mitotic XEEs as before, and then isolated the beads and resuspended them in buffer. We added wild type or mutant RCC1 to the resuspended beads ((A), bottom). Unfortunately, KT proteins on hCENP-A nucleosome arrays were unstable at room temperature after isolation from XEEs, and largely dissociated from the beads within five minutes. By contrast, KT proteins on beads showed minimal changes on ice within five minutes (Fig. S3D). Therefore, we limited their incubation with RCC1 to 5 min on ice, after which we purified the beads again and analyzed the associated proteins by Western blotting ((C)). Compared to beads incubated with buffer only, RCC1WT significantly reduced the levels of Sgo1 and AurB retained. RCC1Ran was similar to RCC1WT for removal of Sgo1 and AurB, but both proteins were insensitive to the addition of RCC1Hist. The capacity of RCC1 to evict Sgo1 and AurB from the defined hCENP-A nucleosome substrate after the removal of most XEE components thus requires RCC1's chromatin-binding affinity but not its enzymatic RanGEF activity. These data are consistent with a mechanism wherein RCC1 competes directly for a binding site on centromeric chromatin ((D)), although we cannot exclude the involvement of other bead-associated proteins.
Discussion
To date, all mechanisms for RCC1's action in different cellular functions have involved Ran-GTP and its capacity to control karyopherin complexes [Citation5]. Changes in RCC1 concentration can alter the composition of mitotic KTs in XEES [Citation6], although the pathway has not been described. Here, we have examined how RCC1 acts in this context. Our findings offer two important insights: First, RCC1-mediated composition changes of replicated KTs are remarkably specific ( & Fig. S1). Exogenous RCC1 selectively displaced Sgo1 and CPC. Outer KT components, including Bub1, Mad2 and Hec1, were markedly less sensitive to RCC1 levels. Second, Ran-GTP did not cause Sgo1 and CPC displacement nor did the loss of RCC1's nucleotide exchange activity strongly impair its capacity to act in this context. On the other hand, RCC1 mutants with reduced capacity to bind chromatin were deficient in Sgo1 and CPC release. These finding suggest a model in which RCC1 physically displaces Sgo1 and CPC ((D)), or in which it promotes other structural re-arrangements leading to their eviction. Remarkably, this suggests a function of RCC1 that does not directly involve its activity as a Ran-GEF.
While unreplicated DSN efficiently nucleate spindles in CSF-XEE, the geometry of their KTs is non-physiological, as are their attachments to spindle MTs. Earlier reports showed that exogenous RCC1 causes mis-localization of many outer KT components from unreplicated DSN [Citation6]. By contrast, equivalent concentrations of exogenous RCC1 caused more limited changes in KT composition on replicated DSN, with specific eviction of a subset of inner KT proteins that includes Sgo1 and CPC ( & Fig. S1). We believe that the earlier observations regarding KT remodeling by RCC1 primarily reflect RCC1's effect on Sgo1 and CPC, and that different responses of outer KT proteins to exogenous RCC1 can be explained for the distinct configurations of unreplicated versus replicated KTs.
RCC1's association to chromatin is dynamic through mitosis in both XEEs and cultured cells [Citation6,Citation7]. RanBP1 controls RCC1 partitioning between cytosol and chromatin in mitosis [Citation8]. In prometaphase, RanBP1 sequesters and inhibits cytosolic RCC1, which is crucial for maintaining a sharp chromatin-oriented Ran-GTP gradient. At anaphase, RanBP1 phosphorylation releases RCC1, allowing it to accumulate on chromatin. RCC1 accumulation on chromatin could facilitate Ran-GTP-dependent events, including NE assembly and reformation of the polarized interphase distribution of Ran-GTP [Citation8]. On the other hand, eviction of Sgo1 and CPC are insensitive to Ran-GTP (). The accumulation of RCC1 on chromatin is coincident with the loss of Sgo1 and CPC from KT, and it is attractive to speculate that RCC1 dynamics may either promote their efficient removal or suppress their reloading, thus ensuring the sequential execution of mitotic events in an irreversible manner.
Both RCC1 and Sgo1 interact directly with histones [Citation4,Citation19]. RCC1 interacts with chromatin through distinct interfaces that contact DNA or Histones, and the N-terminus of RCC1 may further provide a third interface for nucleosome association [Citation16]. RCC1's DNA and Histone binding domains are both important for efficient eviction of inner KT proteins (). In the future, it will be important to understand how RCC1 interacts with centromeric chromatin and particularly whether these interactions are distinct from its association to non-centromeric histones. It is interesting to note that RCC1 was originally identified as a centromeric auto-antigen, CENP-D, raising the possibility that a sub-population of RCC1 may bind to this chromatin domain in a highly regulated and specific fashion [Citation20,Citation21]
In summary, we have demonstrated that elevated levels of RCC1 remodel KTs in XEE through release of a specific subset of inner KT proteins, including Sgo1 and CPC. We have also conclusively demonstrated that remodeling depends only on RCC1's chromatin binding but not its enzymatic RanGEF activity, and that the precise configuration of RCC1 association to chromatin is critical for its capacity to evict Sgo1 and CPC.
Materials and methods
Antibody and Immunodepletion
Antibodies against X. laevis RCC1, Cyclin B2, and Histone H3 were as previously described [Citation8]. Antibodies against X. laevis Sgo1, AurB, Bub1, Mad2, CENP-A have been described elsewhere [Citation22]. Monoclonal antibody 3D11 against hRCC1 was described previously [Citation6]. Antibodies against X. laevis CENP-P (full length), Mad2 (full length), Hec1 (a.a.77-193) were produced in rabbits and purified from serum (Pacific Immunology). Antibodies against X. laevis Cdc27 (BD Bioscience), H. sapiens Histone H2ApThr121 (Active Motif), and H. sapiens CENP-A (USBiological) were purchased from commercial sources. Endogenous RCC1 was immunodepleted from every 100 μl XEEs by incubating with 25 μg anti-xRCC1 antibody crosslinked to 100 μl protein A magnetic beads (Invitrogen).
Immunoblots
SDS-PAGE and membrane transfer were done in a standard procedure. The primary antibodies were diluted in blocking buffer at 1:1,000 (v/v) except for anti-xCyclin B2 that is diluted at 1:500 (v/v). The secondary HRP-linked antibodies against rabbit IgG or mouse IgG (GE Healthcare) were diluted in blocking buffer at 1:5,000 (v/v). SuperSignal Chemiluminescent Substrate (Thermo Scientific) was used for detection. The images were captured with FluorChem R System (Proteinsimple) and quantifications were done using AlphaView software (Proteinsimple).
Recombinant protein expression
The following recombinant proteins were expressed as described previously: X.laevis RCC1 [Citation8]; H.sapiens Ran-GTP, RanT24N, and RanQ69L [Citation23]; H.sapiens RCC1 (wild type and D182 mutant) and RanGAP1 [Citation6]; Histone H2A, H2B, H3, H4, and the hCENP-A/Histone H4 tetramer [Citation10]. Recombinant X.laevis RCC1 mutants were purified the same way as wild type RCC1.
XEEs preparation and use
CSF-arrested X. laevis egg extracts (CSF-XEEs), mitotic XEEs, and demembraned sperm nuclei were prepared as described [Citation8]. To examine KT protein localization, 10,000 DSN/μl were added to XEEs. Sample fixation, purification, and processing for immunofluorescence were as described [Citation22]. Fluorescent images were taken using an ORCA-II CCD camera on a Zeiss Axioskop microscope. The images were acquired and processed with Openlab software. To analyze chromatin-bound proteins, DSN were added to XEE at a final concentration of 10,000 DSN/μl, and incubated at RT for 30 min. Chromatin was isolated and the associated proteins were analyzed by immunoblotting as described [Citation8].
In vitro guanine nucleotide release assay
Recombinant Ran was loaded with [α-32P]GTP (Perkin-Elmer) as described [Citation8]. 1 μM charged Ran-[α-32P]GTP was incubated with 0, 0.1, 0.3, or 1 nM recombinant RCC1 in reaction buffer (50 mM Tris-HCl pH 7.5, 2 mM MgCl2, 100 mM KoAc, 0.5 mg/ml BSA, and 1mM DTT) in a final volume of 25 μl. After 30 min at RT, the reactions were diluted in 500 μl ice cold stop buffer (20 mM Tris-HCl pH 7.5, 25 mM MgCl2, 100 mM NaCl, 1mM DTT) and [α-32P]GTP retention on Ran was measured using a filter binding assay [Citation8]. Triplicate samples were analyzed for each condition. Nucleotide release from the loaded Ran was plotted and analyzed using Prism software. The guanine nucleotide-releasing curve showing mean ± SEM was regressed as one phase decay model.
RCC1 chromatin affinity measurement
After immunodepletion of endogenous RCC1, recombinant RCC1 was added back to XEEs containing chromatin (10,000 DSN/μl) to final concentrations of 6, 20, 60, 200, or 60 μl/ml. Chromatin was isolated and the associated proteins were analyzed and the images were acquired and quantified by quantitative immunoblot using an Odyssey Infrared Imaging System (Li-cor), following instructions from the manufacturer. Chromatin-bound RCC1 levels were plotted against RCC1 concentration in XEEs. Triplicate samples were analyzed for each condition using Prism software. The chromatin-binding curves showing mean ± SEM were regressed as one phase binding model.
Gel filtration assay
Human Histone octamers were extracted from cultured Hela cells as described earlier [Citation24] and mixed with 160bp-dsDNA fragments (Integrated DNA Technologies) containing the Widom 601 DNA sequence [Citation16]. The mixture was incubated at nucleosome assembly buffer (10 mM Tris-HCl pH = 8.0, 1 mM DTT) with 2 M NaCl, 0.6 M NaCl, 0.3 M NaCl, and 25 mM NaCl for 4 hours individually.
2 μM nucleosomes were then mixed with 3 nM × RCC1 in 200 μl buffer (5 mM HEPEs pH = 7.75, 100 mM NaCl, 2 mM MgCl2, 0.5 mg/ml BSA, and 0.1% Tween20) and incubated on ice for 10 min. The mixture was then subjected to Superose 6, 10/300 GL column (GE Healthcare) for separation controlled by AKTA FPLC system (GE Healthcare). The sample was eluted in running buffer (5 mM HEPEs pH = 7.75, 100 mM NaCl, 2 mM MgCl2, and 0.1% Tween20) at a speed of 0.5 ml/min and the eluents were collected in 0.5 fractions. The RCC1 level in each fraction was analyzed by immunoblot.
KT assembly on nucleosome arrays
Nucleosome arrays containing either Histone H3 or Human CENP-A were assembled and conjugated to M-280 Streptavidin beads (Invitrogen) as described [Citation10]. To assemble KTs on beads, 10 μl beads saturated with nucleosome arrays were added into 100 μl CSF-XEEs that were then cycled to mitotic XEEs. Beads were then purified with ice cold washing buffer (5 mM HEPES, pH 7.7, 50 mM KCl, 1 mM MgCl2, 0.2% Triton-X100, 1 μg/ml Okadaic acid, and 1 mM AMPPNP (Roche)). Proteins associated with beads were eluted with 0.1 M Glycine (pH = 2.4) and were analyzed by Western blotting. For analysis of isolated beads ((C)), beads were resuspended in 100 μl washing buffer after from mitotic XEEs, and incubated with recombinant RCC1 for 5 min on ice. Then, the beads were re-purified, eluted, and analyzed using the same procedures.
Disclosure of potential conflict of interest
No potential conflicts of interest were discloded.
SI_file_Zhang_et_al_.docx
Download MS Word (2 MB)Acknowledgements
This study was supported by NICHD project no. HD008740.
Additional information
Funding
Related Research Data
References
- Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Dev Cell. 2014;30:496–508. doi:10.1016/j.devcel.2014.08.016. PMID:25203206
- Gutiérrez-Caballero C, Cebollero LR, Pendás AM. Shugoshins: from protectors of cohesion to versatile adaptors at the centromere. Trends Genet. 2012;28:351–360. doi:10.1016/j.tig.2012.03.003. PMID:22542109
- Carmena M, Wheelock M, Funabiki H, et al. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012;13:789–803. doi:10.1038/nrm3474. PMID:23175282
- Watanabe Y. Temporal and spatial regulation of targeting aurora B to the inner centromere. Cold Spring Harb Symp Quant Biol. 2010;75:419–423. doi:10.1101/sqb.2010.75.035. PMID:21447816
- Clarke PR, Zhang C. Spatial and temporal coordination of mitosis by Ran GTPase. Nat Rev Mol Cell Biol. 2008;9:464–477. doi:10.1038/nrm2410. PMID:18478030
- Arnaoutov A, Dasso M. The Ran GTPase regulates kinetochore function. Dev Cell. 2003;5:99–111. doi:10.1016/S1534-5807(03)00194-1. PMID:12852855
- Hutchins JRA, Moore WJ, Hood FE, et al. Phosphorylation regulates the dynamic interaction of RCC1 with chromosomes during mitosis. Curr Biol. 2004;14:1099–1104. doi:10.1016/j.cub.2004.05.021. PMID:15203004
- Zhang MS, Arnaoutov A, Dasso M. RanBP1 governs spindle assembly by defining mitotic Ran-GTP production. Dev Cell. 2014;31:393–404. doi:10.1016/j.devcel.2014.10.014. PMID:25458009
- Milks KJ, Moree B, Straight AF. Dissection of CENP-C-directed centromere and kinetochore assembly. Mol Biol Cell. 2009;20:4246–4255. doi:10.1091/mbc.E09-05-0378. PMID:19641019
- Guse A, Fuller CJ, Straight AF. A cell-free system for functional centromere and kinetochore assembly. Nat Protoc. 2012;7:1847–1869. doi:10.1038/nprot.2012.112. PMID:23018190
- Minshull J, Sun H, Tonks NK, et al. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 1994;79:475–486. doi:10.1016/0092-8674(94)90256-9. PMID:7954813
- Boiarchuk EI, Nikol'skiĬ NN, Dasso M, et al. [Assembly of correct kinetochore architecture in Xenopus laevis egg extract requires transition of sperm DNA through interphase]. Tsitologiia. 2007;49:502–511. PMID:17802748
- Kawashima SA, Yamagishi Y, Honda T, et al. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science. 2010;327:172–177. doi:10.1126/science.1180189. PMID:19965387
- Stewart M, Kent HM, McCoy AJ. The structure of the Q69L mutant of GDP-Ran shows a major conformational change in the switch II loop that accounts for its failure to bind nuclear transport factor 2 (NTF2). J Mol Biol. 1998;284:1517–1527. doi:10.1006/jmbi.1998.2204. PMID:9878368
- Azuma Y, Renault L, García-Ranea JA, et al. Model of the ran-RCC1 interaction using biochemical and docking experiments. J Mol Biol. 1999;289:1119–1130. doi:10.1006/jmbi.1999.2820. PMID:10369786
- Makde RD, England JR, Yennawar HP, et al. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010;467:562–566. doi:10.1038/nature09321. PMID:20739938
- Renault L, Kuhlmann J, Henkel A, et al. Structural basis for guanine nucleotide exchange on Ran by the regulator of chromosome condensation (RCC1). Cell. 2001;105:245–255. doi:10.1016/S0092-8674(01)00315-4. PMID:11336674
- Furuta M, Hori T, Fukagawa T. Chromatin binding of RCC1 during mitosis is important for its nuclear localization in interphase. Mol Biol Cell. 2016;27:371–381. doi:10.1091/mbc.E15-07-0497. PMID:26564799
- Nemergut ME, Mizzen CA, Stukenberg T, et al. Chromatin docking and exchange activity enhancement of RCC1 by histones H2A and H2B. Science. 2001;292:1540–1543. doi:10.1126/science.292.5521.1540. PMID:11375490
- Bischoff FR, Maier G, Tilz G, et al. A 47-kDa human nuclear protein recognized by antikinetochore autoimmune sera is homologous with the protein encoded by RCC1, a gene implicated in onset of chromosome condensation. Proc Natl Acad Sci USA. 1990;87:8617–8621. doi:10.1073/pnas.87.21.8617. PMID:2236072
- Earnshaw WC, Rattner JB. The use of autoantibodies in the study of nuclear and chromosomal organization. Methods Cell Biol. 1991;35:135–175. doi:10.1016/S0091-679X(08)60572-5. PMID:1723479
- Boyarchuk Y, Salic A, Dasso M, et al. Bub1 is essential for assembly of the functional inner centromere. J Cell Biol. 2007;176:919–928. doi:10.1083/jcb.200609044. PMID:17389228
- Dasso M, Seki T, Azuma Y, et al. A mutant form of the Ran/TC4 protein disrupts nuclear function in Xenopus laevis egg extracts by inhibiting the RCC1 protein, a regulator of chromosome condensation. EMBO J. 1994;13:5732–5744. PMID:7988569
- Rodriguez-Collazo P, Leuba SH, Zlatanova J. Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic Acids Res. 2009;37:e81–1. doi:10.1093/nar/gkp273. PMID:19443446