
ABSTRACT
Although the p53 transcription factor has a well-established role in tumor suppression, little is known about how the non-coding targets of p53 mediate its tumor suppression function. Analysis of ncRNAs regulated by p53 revealed Neat1 as a direct p53 target gene. Neat1 has physiological roles in the development and differentiation of the mammary gland and corpus luteum, but its roles in cancer have been conflicting. To unequivocally understand Neat1 function in cancer, we used Neat1 null mice. Interestingly, we found that Neat1 deficiency promotes transformation both in oncogene-expressing fibroblasts and in a mouse model for pancreatic cancer. Specifically, Neat1 loss in the pancreas results in the enhanced development of preneoplastic lesions associated with dampened expression of differentiation genes. While the exact mechanisms underlying tumor suppression are unknown, there are several described mechanisms that may be responsible for Neat1-mediated tumor suppression. Collectively, these findings suggest that Neat1 enforces differentiation to suppress pancreatic cancer.
Introduction
The p53 transcription factor plays a critical role in tumor suppression, as illustrated by the high incidence of p53 mutations in a variety of human tumor types and the completely penetrant cancer phenotype of p53 null mice [Citation1,Citation2]. p53 is a cellular stress sensor, activated by signals such as DNA damage and oncogene expression. Upon activation, p53 can induce cell cycle arrest, which allows damaged cells time to repair their DNA before re-entering the cell cycle, thus promoting genome integrity and cell survival. Alternatively, p53 can respond to stress signals by triggering apoptosis to eliminate damaged or hyperproliferative cells. In recent years, many additional processes potentially relevant to tumor suppression have been found to be regulated by p53, such as metabolic reprogramming, differentiation, and migration [Citation3]. The ability of p53 to suppress cancer depends on its function as a transcriptional activator, as clearly demonstrated using mouse models, although the transcriptional targets through which it suppresses cancer remain elusive [Citation4–Citation6]. p53 binds specific DNA response elements throughout the genome and regulates a panoply of genes, including protein-coding genes and non-coding RNAs (ncRNAs). Although functional studies of p53-regulated genes have largely focused on protein-coding genes, a cadre of ncRNAs, including several microRNAs, have also been functionally analyzed and shown to contribute to the p53 pathway in different ways. These ncRNAs include large intergenic ncRNAs (lincRNAs), a class of ncRNAs that have received significant attention for their diverse mechanisms in regulating different biological processes, including cancer. These p53-regulated lincRNAs, which modulate p53 stability, activity and responses, include: LincRNAp21 [Citation7,Citation8], PANDA [Citation9], Pint [Citation10], PVT1 [Citation11,Citation12], PR-lncRNA-1 and 10 [Citation13], PINCR [Citation14], TUG1 [Citation15,Citation16], LED [Citation17], TRINGS [Citation18], DINO [Citation19], and PURPL [Citation20],(). Only loc285194 and LincRNAp21 have been reported to function in tumor suppression, as expression of either suppresses xenograft tumor growth in mice [Citation21,Citation22]. Since these initial discoveries, the next-generation sequencing revolution has continued to lead to the identification of novel lincRNAs that could be important for understanding p53 function in tumor suppression [Citation23]. In fact, analysis of p53 ChIP-sequencing and RNA-sequencing experiments in p53-proficient and deficient cells was essential for the recent identification of the Neat1 lincRNA as a p53 target gene [Citation24–Citation28], the subject of this commentary.
Table 1. LincRNAs directly regulated by p53 with functions potentially relevant to p53-mediated tumor suppression
Neat1 was initially described as a Virus-Inducible ncRNA (VINC) upregulated in the mouse brain upon infection with Japanese encephalitis or Rabies viruses [Citation29]. Subsequent studies demonstrated that Neat1 is also upregulated in response to infection of other viruses, such as HIV, Influenza virus and herpesvirus, and is involved in the induction of innate immune responses, thereby limiting viral replication [Citation30–Citation32]. Shortly thereafter, another study described two ubiquitously expressed nuclear-enriched ncRNAs, which were named NEAT1 and NEAT2 (Nuclear Enriched Autosomal noncoding Transcripts; NEAT2 is also known as MALAT1) [Citation33]. This study further demonstrated that NEAT1 has two variant transcripts, one short (~3.7 KB, named NEAT1_1) and one much longer variant (~23 KB, named NEAT1_2). Moreover, NEAT1 localizes to subnuclear structures called paraspeckles – RNA- and protein-containing structures proposed to control the expression of protein-coding genes by retaining their cognate A-to-I edited RNAs in the nucleus [Citation34]. Interestingly, Neat1 is essential for the formation of paraspeckles [Citation35] and both Neat1 and paraspeckles are specific to mammalian cells [Citation36], suggesting that this type of nuclear compartmentalization might represent a strategy that is needed for the complex regulation of gene expression occurring in mammals.
Physiological roles for Neat1
Our understanding of Neat1 has come in part from investigating its physiological functions in mice. Initial studies of Neat1 null mice revealed no obvious phenotypes, suggesting that Neat1 is dispensable for normal development [Citation37]. However, subsequent studies showed that Neat1 null females display compromised lactation, associated with reduced mammary epithelial cell proliferation and defective lobular-alveolar development [Citation38]. In addition, Neat1 null mice exhibit defects in ductal and branching morphogenesis, indicating a role for Neat1 in the proper development of mammary ducts [Citation38]. Neat1 deficiency is also associated with a stochastic impaired ability of female mice to get pregnant [Citation39]. This phenotype is attributable to defective formation of the corpus luteum – a temporary endocrine structure involved in the establishment of pregnancy – and diminished progesterone levels. This corpus luteum defect may be explained by the requirement for Neat1 for the proper expression of genes necessary for corpus luteum differentiation, such as Star. These findings are in keeping with previous observations suggesting a potential role for Neat1 in differentiation in various contexts. For example, NEAT1 is expressed in many cell types, with the exception of embryonic stem cells, where its expression is only detected upon differentiation [Citation40]. NEAT1 is also upregulated during differentiation of neurons, glia, myeloid cells, and muscle, further supporting a role for NEAT1 in cellular differentiation [Citation41–Citation44].
The role of Neat1 in tumorigenesis
Beyond analysis of the physiological functions of Neat1, additional studies have focused on the role of Neat1 during cancer development. Following initial observations that NEAT1 is elevated in a variety of human cancers relative to normal tissue counterparts [Citation45], suggesting a pro-tumorigenic function, analysis of human cancer data focused on correlating NEAT1 expression with cancer outcome. Higher levels of NEAT1 were connected to worse prognosis in a range of human cancers, such as colorectal, laryngeal and gastric cancers, as well as glioma, suggesting that NEAT1 could be acting as an oncogene in these cancers [Citation46–Citation49]. Some functional studies in vitro using cancer cell lines from liver, breast, prostate and nasopharyngeal tumors also suggested that NEAT1 expression could enhance cell proliferation, cell survival, cell invasion, epithelial-mesenchymal transition (EMT), and subcutaneous tumor growth [Citation25,Citation48,Citation50–Citation55]. Perhaps the most compelling evidence to support the oncogenic activity of NEAT1 came from a study of Neat1 knockout mice subjected to a DMBA-TPA skin carcinogenesis protocol [Citation25]. In this study, Neat1 deficiency was shown to confer resistance to chemically-induced skin cancer. This oncogenic function was attributed to a role for Neat1 in promoting ATR signaling in the face of replication stress, preventing the accumulation of DNA double strand breaks and consequent death of incipient cancer cells.
Previous studies have also suggested that NEAT1 may have tumor suppressor activity in some contexts. First, NEAT1 expression was found to be downregulated in lung, liver, esophageal, nasopharyngeal, and retinal cancers, as well as acute promyelocytic leukemias, relative to normal tissue counterparts [Citation27,Citation44,Citation56,Citation57]. Second, NEAT1 overexpression can reduce cell proliferation in lung cancer and osteosarcoma cell lines upon treatment with nutlin or Adriamycin [Citation27]. Third, high-level NEAT1 expression predicted better overall survival in nasopharyngeal cancer, and NEAT1 overexpression radically decreased nasopharyngeal cancer cell growth in subcutaneous tumor studies, through a mechanism that involves the inhibition of miR-101-3p [Citation57]. Finally, recent studies have shown that a high frequency of point mutations and deletions are detected in the NEAT1 promoter in both liver and breast cancers [Citation50,Citation58], and that mutations found in the NEAT1 promoter in breast cancers are typically associated with decreased expression of NEAT1. Although the point mutations described in breast cancer have not yet been shown to involve the p53 binding site, the deletions typically involve the p53 response element. In addition, NEAT1 was suggested to promote therapeutic responses, as the successful treatment of acute promyelocytic leukemias with all-trans retinoic acid (ATRA), which induces differentiation and blocks tumor cell propagation, depends on the presence of NEAT1 [Citation44].
Taken together, some of these studies have supported a role for NEAT1 in promoting cancer, while other studies have demonstrated a role for NEAT1 in suppressing cancer. These apparently contradictory results can be reconciled by a couple of different explanations. First, it may be that the two NEAT1 isoforms have different cellular functions and many of the aforementioned studies may not have studied the specific isoforms involved in the context examined. Indeed, increased NEAT1_2 expression correlates with better survival in colorectal cancer patients, while the opposite is observed with the isoform NEAT1_1 [Citation59]. Moreover, the knockdown of NEAT1_1 in colorectal cancer cell lines attenuated invasiveness and cell growth, while the knockdown of NEAT1_2 enhanced cell growth in the same cell lines. Second, it may be that NEAT1’s mechanism of action differs according to the cell type or context, with it acting as a tumor suppressor in some settings and an oncogene in others. It is also possible that NEAT1 might have different roles at different stages of tumorigenesis, initially suppressing tumorigenesis, and later promoting tumor development. This paradigm is not uncommon, as in the case of β-catenin which inhibits early pancreatic cancer lesions but promotes the development of later stage lesions [Citation60], or the apoptosis-promoting protein Perp which is necessary at early steps of skin papilloma development, but opposes carcinogenesis at later stages [Citation61]. Finally, many of these studies were correlative rather than functional, highlighting the necessity for functional studies.
Using Neat1 knockout mice to study tumorigenesis
To unequivocally understand the role of Neat1 in cancer development, we recently used Neat1 null mice as a clean genetic model to study the consequences of complete Neat1 deficiency for tumorigenesis. We first derived primary mouse embryonic fibroblasts (MEFs) from these mice to assess the contribution of Neat1 to transformation suppression in a classic oncogene-expressing fibroblast model, in which p53 plays a central role. In this model, E1A;HrasG12V oncogene-expressing mouse embryonic fibroblasts (MEFs) with intact p53 form colonies inefficiently in transformation assays, whereas E1A; HrasG12V;p53-/- MEFs efficiently form colonies. Interestingly, we found that E1A;HrasG12V MEFs null for Neat1 exhibit greatly increased colony formation, to levels that rival the enhanced colony formation seen upon p53 loss [Citation24]. We further established the role of Neat1 in transformation suppression in vivo, in a subcutaneous xenograft model where we found the E1A;HrasG12V-expressing fibroblasts formed tumors much faster when null for Neat1. Next we sought to examine the role of Neat1 in transformation suppression in epithelial cancers, which constitute the vast majority of human cancers. RNA-sequencing analysis of premalignant pancreatic cancer lesions showed that Neat1 expression is p53-dependent in these cells, and we therefore focused on pancreatic cancer development. Consistent with a transformation suppression role in these cells, we found that Neat1_1 overexpression sufficed to inhibit transformation of p53 null pancreatic cancer cells. To establish the significance of this role in vivo, we analyzed the consequences of Neat1 deficiency in an autochthonous mouse model for pancreatic cancer driven by Cre-activated KrasG12D expression in the pancreas [Citation62]. In this model, either the acinar cells (epithelial cells responsible for producing digestive enzymes) or the ductal cells (epithelial-cells lining the ducts that deliver digestive enzymes into the duodenum) of the exocrine pancreas can undergo dedifferentiation and cellular transformation upon constitutive Kras activation, giving rise to premalignant lesions known as pancreatic intraepithelial neoplasias (PanINs) and cystic and intraductal papillary mucinous neoplasms (IPMNs), respectively [Citation63,Citation64]. Strikingly, KrasG12D-expressing Neat1 deficient mice succumbed to enhanced development of pancreatic neoplasias, including PanINs and IPMN-like precursor lesions, relative to Neat1-expressing mice () [Citation24]. While we did not observe cancer in the time frame examined, these premalignant lesions may ultimately evolve into metastatic pancreatic cancer. Together, these studies revealed that Neat1 has an essential role in suppressing the transformation of normal pancreatic acinar and ductal cells into premalignant lesions. Our study was thus the first to demonstrate a transformation suppression role for Neat1 in vivo, in a well-established genetically engineered mouse model for cancer.
Figure 1. The role of Neat1 in suppressing transformation in pancreatic cancer
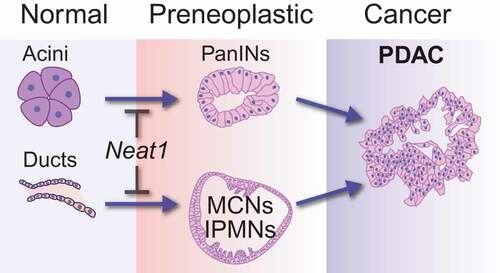
To reveal the underlying mechanisms for Neat1 transformation suppression, we analyzed genome-wide expression in E1A;HrasG12V-expressing wild-type or Neat1 null fibroblasts by RNA-sequencing. We then compared the gene expression profiles of these Neat1-proficient and deficient cells, which led to the conclusion that Neat1 controls the transcriptional output of 1305 genes (q-value: 0.005, FDR: 0.5%). Interestingly, we observed that many of these genes are also regulated by Neat1 in the pancreas of mice harboring KrasG12D mutations, suggesting that Neat1’s global control of transcription is a general effect observed in different cell types. Among the genes downregulated in Neat1 null cells, there was an enrichment of genes associated with different developmental programs, including nervous system development, axon guidance, and pancreas development. The genes involved in pancreas development included Bhlha15 (also known as Mist1) and Sox9. While the former has been shown to be essential for late-stage differentiation of acinar cells, the latter has a general role in pancreas development and is known as a transcription factor that maintains ductal cell identity. The downregulation of Bhlha15 or Sox9 in the pancreas of Neat1-deficient mice could cause dedifferentiation and increased formation of PanINs or IPMN-like lesions, respectively. Our data suggest that Neat1 acts as a tumor suppressor by safeguarding the expression of genes involved in cellular differentiation, thus opposing de-differentiation events driven by oncogenic drivers such as mutant Kras.
Potential mechanisms for NEAT1 action in tumor suppression
The exact mechanisms by which NEAT1 might regulate gene expression to suppress transformation are unclear, but could relate to any of several different molecular functions ascribed to NEAT1, all ultimately impinging upon gene expression (). As mentioned briefly above, one of the first functions associated with paraspeckles, and by association with NEAT1, was the ability to post-transcriptionally modulate the expression of genes that undergo RNA editing (A-to-I), through the nuclear retention of edited RNAs in paraspeckles [Citation34,Citation36,Citation40]. Interestingly, this mechanism of action is reversible, and upon stress, the edited sequence can be cleaved from the RNAs, allowing release from the nucleus, delivery to the cytoplasm, and expression of the regulated gene. NEAT1 was also found to interact with the splicing machinery and the Drosha-DGCR8 Microprocessor, regulating the maturation of pre-mRNAs and pri-miRNAs [Citation65–Citation68], respectively, thereby controlling processes that rely on alternative splicing or on miRNA-regulated gene expression programs. Yet another way that NEAT1 has been shown to change the transcriptional output of the cell is through the retention of proteins such as transcription factors in paraspeckles [Citation69]. Interestingly, the retention of one single protein, the transcriptional and splicing regulator SFPQ, in paraspeckles via interaction with NEAT1 impedes the ability of SFPQ to bind the chromatin of target genes, leading to changes in the transcriptional output, such as the repression of ADAR2B and the induction of IL8 [Citation31,Citation69]. There is also evidence that NEAT1 can globally directly interact with the chromatin of hundreds of active genes, though little is still known about the impact of these interactions [Citation65]. One hypothesis is that NEAT1 could bring together proteins from the paraspeckles and other subnuclear structures to the chromatin of active genes to regulate the expression of these genes [Citation65]. NEAT1 interactions with the chromatin might direct changes in the epigenetic landscape of the cells, as NEAT1 has been shown to alter chromatin marks such as histone H3K4 trimethylation and histone H3K9 acetylation in specific genes [Citation54]. NEAT1 could also play a role in the control of chromatin remodeling. Proteins from the SWI/SNF chromatin remodeling complex were shown to be major components of the paraspeckles [Citation70], where they participate in the assembly of the paraspeckles, in a nucleosome-remodeling activity-independent fashion. This unexpected interaction begs the question of whether or not NEAT1 could alter chromatin organization by bringing these proteins to the chromatin of active genes or by retaining them at the paraspeckles. Interestingly, in our studies we observed that Neat1 deficiency in fibroblasts decreases the expression of chromatin remodeling genes such as Smarca1, Smarcc2 and Arid1a, which encode proteins from the SWI/SNF complex, reinforcing the connection between NEAT1 and the SWI/SNF complex [Citation24].
Figure 2. Mechanisms of Neat1 action
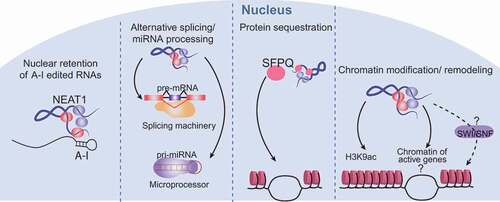
Collectively, these observations suggest a variety of potential mechanisms by which NEAT1 might impact tumor development. It is possible that NEAT1 suppresses tumors by impinging on gene expression through multiple different mechanisms at the same time or that only a subset of these mechanisms is essential for its function as a tumor suppressor. Furthermore, there may be tissue-specific differences in mechanisms of NEAT1 action. Dissecting the molecular mechanisms that NEAT1 employs in tumor suppression will ultimately expand our understanding of how lincRNAs contribute to tumorigenesis.
Summary and future perspectives
Our studies here using genetic mouse models have revealed a clear role for Neat1 as a tumor suppressor, and suggest that Neat1 loss may facilitate cellular transformation by promoting dedifferentiation. These observations suggest in turn that one aspect of p53 function is through activation of Neat1 expression and stimulation of a differentiation program. Many questions remain, however. For example, is Neat1’s role as a tumor suppressor tissue-specific? The continued use of a clean genetic mouse model for Neat1 deficiency in in vivo studies will be essential for determining Neat1’s role in tumorigenesis in different tissues. How does Neat1 act at the molecular level to suppress cancer? The use of cutting-edge genomic techniques such as ChiRP-seq – a technique to detect RNA-chromatin interactions – and CLIP-seq – a genome-wide technique to map protein/RNA interactions – combined with RNA-seq, in a tumor suppression context, will help address this question by describing how interactions between Neat1, chromatin and RNA binding proteins form key regulatory networks that impact gene expression. Such future investigations will unravel the exact mechanisms of action of Neat1 in tumor suppression, as well as the key proteins and chromatin interactions involved in this process.
Acknowledgments
We thank N. Raj, B.M. Flowers, and A. Kaiser for critical discussions and reading of the manuscript. We apologize to those whose work we could not cite due to space constraints.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008.
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431.
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370.
- Brady CA, Jiang D, Mello SS, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583.
- Jiang D, Brady CA, Johnson TM, et al. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci. 2011;108:17123–17128.
- Mello SS, Valente LJ, Raj N, et al. A p53 super-tumor suppressor reveals a tumor suppressive p53-Ptpn14-Yap axis in pancreatic cancer. Cancer Cell. 2017;32:460–73.e6.
- Dimitrova N, Zamudio JR, Jong RM, et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell. 2014;54:777–790.
- Huarte M, Guttman M, Feldser D, et al. A large intergenic noncoding RNA Induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419.
- Hung T, Wang Y, Lin MF, et al. Extensive and coordinated transcription of noncoding RNAs within cell cycle promoters. Nat Genet. 2011;43:621–629.
- Marín-Béjar O, Marchese FP, Athie A, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14:1–17.
- Barsotti AM, Beckerman R, Laptenko O, et al. p53-Dependent induction of PVT1 and miR-1204. J Biol Chem. 2012;287:2509–2519.
- Tseng YY, Moriarity BS, Gong W, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86.
- Sánchez Y, Segura V, Marín-Béjar O, et al. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun. 2014;5:5812.
- Chaudhary R, Gryder B, Woods WS, et al. Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to Matrin 3. eLife. 2017;6:e23244.
- Zhang E, Yin D, Sun M, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014;5:e1243.
- Fan S, Yang Z, Ke Z, et al. Downregulation of the long non-coding RNA TUG1 is associated with cell proliferation, migration, and invasion in breast cancer. Biomed Pharmacother. 2017;95:1636–1643.
- Leveille N, Melo CA, Rooijers K, et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun. 2015;6:6520.
- Khan MR, Xiang S, Song Z, et al. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. Embo J. 2017;36:3483–3500.
- Schmitt AM, Garcia JT, Hung T, et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet. 2016;48:1370–1376.
- Li XL, Subramanian M, Jones MF, et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 2017;20:2408–2423.
- Liu Q, Huang J, Zhou N, et al. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013;41:4976–4987.
- Wang J, Lei ZJ, Guo Y, et al. miRNA-regulated delivery of lincRNA-p21 suppresses beta-catenin signaling and tumorigenicity of colorectal cancer stem cells. Oncotarget. 2015;6:37852–37870.
- Kawaji H, Severin J, Lizio M, et al. Update of the FANTOM web resource: from mammalian transcriptional landscape to its dynamic regulation. Nucleic Acids Res. 2011;39:D856–D60.
- Mello SS, Sinow C, Raj N, et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017;31:1095–1108.
- Adriaens C, Standaert L, Barra J, et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. 2016;22:861–868.
- Blume CJ, Hotz-Wagenblatt A, Hullein J, et al. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia. 2015;29:2015–2023.
- Idogawa M, Ohashi T, Sasaki Y, et al. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int J Cancer. 2017;140:2785–2791.
- Botcheva K, McCorkle SR, McCombie WR, et al. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011;10:4237–4249.
- Saha S, Murthy S, Rangarajan PN. Identification and characterization of a virus-inducible non-coding RNA in mouse brain. J Gen Virol. 2006;87:1991–1995.
- Morchikh M, Cribier A, Raffel R, et al. HEXIM1 and NEAT1 long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immune response. Mol Cell. 2017;67:387–99.e5.
- Imamura K, Imamachi N, Akizuki G, et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell. 2014;53:393–406.
- Zhang Q, Chen CY, Yedavalli VS, et al. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio. 2013;4:e00596–00512.
- Hutchinson JN, Ensminger AW, Clemson CM, et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39.
- Prasanth KV, Prasanth SG, Xuan Z, et al. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–263.
- Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–726.
- Fox AH, Lamond AI. Paraspeckles. Cold Spring Harb Perspect Biol. 2010;2:a000687–a.
- Nakagawa S, Naganuma T, Shioi G, et al. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol. 2011;193:31–39.
- Standaert L, Adriaens C, Radaelli E, et al. The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA. 2014;20:1844–1849.
- Nakagawa S, Shimada M, Yanaka K, et al. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development. 2014;141:4618–4627.
- Chen -L-L, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell. 2009;35:467–478.
- Sunwoo H, Dinger ME, Wilusz JE, et al. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19:347–359.
- Lehnert SA, Reverter A, Byrne KA, et al. Gene expression studies of developing bovine longissimusmuscle from two different beef cattle breeds. BMC Dev Biol. 2007;7:1–13.
- Mercer TR, Qureshi IA, Gokhan S, et al. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 2010;11:1–15.
- Zeng C, Xu Y, Xu L, et al. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer. 2014;14:1–7.
- Yang C, Li Z, Li Y, et al. Long non-coding RNA NEAT1 overexpression is associated with poor prognosis in cancer patients: a systematic review and meta-analysis. Oncotarget. 2017;8:2672–2680.
- Li Y, Li Y, Chen W, et al. NEAT expression is associated with tumor recurrence and unfavorable prognosis in colorectal cancer. Oncotarget. 2015;6:27641–27650.
- He C, Jiang B, Ma J, et al. Aberrant NEAT1 expression is associated with clinical outcome in high grade glioma patients. APMIS. 2016;124:169–174.
- Wang P, Wu T, Zhou H, et al. Long noncoding RNA NEAT1 promotes laryngeal squamous cell cancer through regulating miR-107/CDK6 pathway. J Exp Clin Cancer Res. 2016;35:1–11.
- Ma Y, Liu L, Yan F, et al. Enhanced expression of long non-coding RNA NEAT1 is associated with the progression of gastric adenocarcinomas. World J Surg Oncol. 2016;14:1–6.
- Fujimoto A, Furuta M, Totoki Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500–509.
- Choudhry H, Albukhari A, Morotti M, et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene. 2015;34:4482–4490.
- Fang L, Sun J, Pan Z, et al. Long non-coding RNA NEAT1 promotes hepatocellular carcinoma cell proliferation through the regulation of miR-129-5p-VCP-IkappaB. Am J Physiol Gastrointest Liver Physiol. 2017;313:G150–g6.
- Lu Y, Li T, Wei G, et al. The long non-coding RNA NEAT1 regulates epithelial to mesenchymal transition and radioresistance in through miR-204/ZEB1 axis in nasopharyngeal carcinoma. Tumor Biol. 2016;37:11733-11741.
- Chakravarty D, Sboner A, Nair SS, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014;5:5383.
- Zheng X, Zhang Y, Liu Y, et al. HIF-2alpha activated lncRNA NEAT1 promotes hepatocellular carcinoma cell invasion and metastasis by affecting the epithelial-mesenchymal transition. J Cell Biochem. 2017;119:3247-3256.
- Gibb EA, Vucic EA, Enfield KSS, et al. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6:e25915.
- Wang Y, Wang C, Chen C, et al. Long non-coding RNA NEAT1 regulates epithelial membrane protein 2 expression to repress nasopharyngeal carcinoma migration and irradiation-resistance through miR-101-3p as a competing endogenous RNA mechanism. Oncotarget. 2017;8:70156–70171.
- Rheinbay E, Parasuraman P, Grimsby J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55–60.
- Wu Y, Yang L, Zhao J, et al. Nuclear-enriched abundant transcript 1 as a diagnostic and prognostic biomarker in colorectal cancer. Mol Cancer. 2015;14:1–12.
- Morris J, Cano DA, Sekine S, et al. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–520.
- Marques MR, Horner JS, Ihrie RA, et al. Mice lacking the p53/p63 target gene Perp are resistant to papilloma development. Cancer Res. 2005;65:6551–6556.
- Hingorani SR, Petricoin Iii EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450.
- Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302.
- Matthaei H, Schulick RD, Hruban RH, et al. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8:141–150.
- West JA, Davis CP, Sunwoo H, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014;55:791–802.
- Cooper D, Carter G, Li P, et al. Long non-coding RNA NEAT1 associates with SRp40 to temporally regulate PPARγ2 splicing during adipogenesis in 3T3-L1 cells. Genes. 2014;5:1050–1063.
- Cooper DR, Carter G, Li P, et al. Long non-coding RNA NEAT 1 & 2 regulates phosphorylation of SR proteins and PKCβII splicing during 3T3 L1 adipogenesis. FASEB J. 2012;26:941.942.
- Jiang L, Shao C, Wu QJ, et al. NEAT1 scaffolds RNA-binding proteins and the microprocessor to globally enhance pri-miRNA processing. Nat Struct Mol Biol. 2017;24:816–824.
- Hirose T, Virnicchi G, Tanigawa A, et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol Biol Cell. 2014;25:169–183.
- Kawaguchi T, Tanigawa A, Naganuma T, et al. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc Natl Acad Sci. 2015;112:4304–4309.