
ABSTRACT
Small RNAs play an important role in gene regulatory networks. The gene suppressive effect of small RNAs was previously the dominant focus of studies, but during the recent decade, small RNA-induced gene activation has been reported and has become a notable gene manipulation technique. In this study, a putative tumor suppressor, INTS6, was activated by introducing a promoter-targeted small RNA (dsRNA-915) into castration-resistant prostate cancer (CRPC) cells. Unique dynamics associated with the gene upregulation phenomenon was observed. Following gene activation, cell proliferation and motility were suppressed in vitro. Downregulation of Wnt/β-catenin signaling was observed during the activation period, and the impairment of β-catenin degradation reversed the tumor suppressor effects of INTS6. These results suggest the potential application of small activating RNAs in targeted gene therapy for CRPC.
Introduction
Prostate cancer (PCa) is the second most common cancer and the fifth leading cause of cancer death in males worldwide [Citation1]. Hormonal therapy, which mainly suppresses androgen signaling, remains the first choice for advanced prostate cancer and is strongly recommended as an adjuvant therapy for high- and intermittent-risk disease [Citation2]. Although a relatively high primary response rate is observed, almost all patients experience disease progression into castration-resistant prostate cancer (CRPC) over a period of 2 to 3 years, with a poor prognosis despite newer second-line cytotoxic chemotherapy and endocrine therapies [Citation3–Citation5].
Gene activation by small RNAs was first reported by Li in 2006 [Citation6], and this phenomenon was termed RNA activation (RNAa) in contrast to RNAi. Small double-strand activating RNAs (saRNAs) targeting select promoter regions successfully activated the human genes E-cadherin, p21WAF1/CIP1 (p21), and VEGF at the transcription level. Following this study, a few studies reported that genes could be activated by small RNAs, including microRNAs, in a variety of mammalian cells [Citation7–Citation11]. These results suggested a general activating effect of small RNAs in cell biology. Although the mechanism of RNAa action has not been well elucidated, the activation course and specific epigenetic modulation of targeted genes distinguish RNAa as a unique method of gene regulation [Citation6,Citation7,Citation12].
RNAa is a novel strategy for targeted gene therapies. E-cadherin and p21 are tumor suppressor genes that are susceptible to RNAa. Activation of these genes by small RNAs successfully suppressed different cancer cells both in vitro and in vivo[Citation13–Citation15]. VEGF plays an important role in angiogenesis. Upregulation of VEGF by promoter-targeted shRNA increased the local vascularity and blood flow in an ischemic mouse model [Citation10]. In another study, dsRNAs complementary to the promoter sequence of the LDLR (low-density lipoprotein receptor) gene activated the targeted gene and increased LDLR expression on the surface of liver cells, indicating a potential application in hyperlipidemia therapy [Citation16].
Integrator complex subunit 6 (INTS6), which was identified in 1999, is localized on chromosome 13q14 [Citation17,Citation18]. As frequent loss of heterozygosity (LOH) occurs in this area in malignant tumors, INTS6 may be a putative tumor suppressor gene [Citation17–Citation19]. In this study, we activated INTS6 gene expression in CRPC cells with a promoter-targeted small RNA. Following the activation of INTS6, cell proliferation and motility were suppressed. In addition, the downregulation of β-catenin was found to be crucial for the tumor suppressor effect of INTS6.
Results
Frequent downregulation of INTS6 was detected by immunohistochemistry staining of prostate cancer tissues
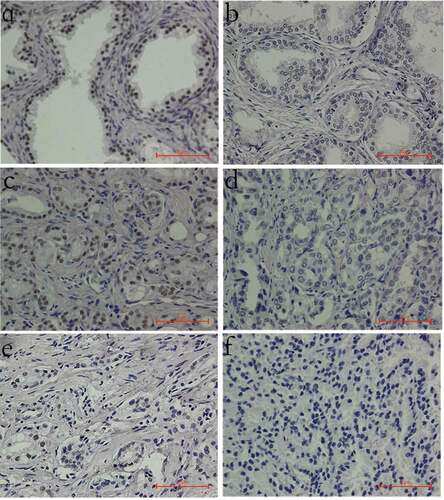
LOH involving INTS6 was reported in 21 ~ 48% of PCa tissues[Citation18,Citation20,Citation21]. A relatively lower mRNA level of INTS6 has also been detected in PCa cell lines [Citation22,Citation23]. However, INTS6 protein levels in PCa tissues have yet to be well detected. In this study, INTS6 protein levels were detected by immunohistochemistry in a tissue array containing 28 pairs of malignant and benign prostate tissues. The INTS6 protein mainly localized to the nucleus. Decreased staining was observed in 3.6% (1/28) of benign tissues and 25% (7/28) of cancer tissues, whereas 3.6% (1/28) of benign tissues and 10.7% (3/28) of cancer tissues showed a negative staining pattern (). A significant difference in staining intensity was observed between the benign and malignant tissues, and this difference was independent of patient age (). The Gleason score (GS) is an independent predictor of survival and treatment outcome in patients with PCa. When the GS increased, the reverse trend was observed in INTS6 staining intensity in PCa tissues, with borderline significance (P = 0.051, Table S1).
Table 1. Comparison of the staining intensity between benign and malignant prostate tissues
Figure 1. INTS6 protein staining patterns in prostate tissues (magnification, 400×). (a) Positive INTS6 staining in the nucleus of most benign prostate tissues. (b) Decreased INTS6 staining in a benign prostate tissue sample. (c) Positive INTS6 staining in a prostate cancer tissue sample. (d, e) Decreased staining intensity or a decreased proportion of positive nuclei in prostate cancer tissue. (f) Negative INTS6 staining in a prostate cancer tissue sample
Promoter-targeted small RNA activated INTS6 expression in human cell lines
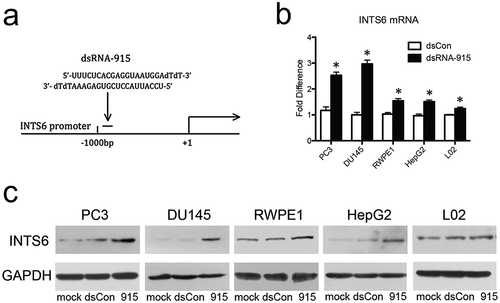
According to the protocol described by Li [Citation6] and Yang [Citation11], several small double-strand RNAs (dsRNAs) homologous to the promoter region of the INTS6 gene were designed. These small RNA candidates were transfected into CRPC cells (PC3 and DU145) using a liposome capsule, and INTS6 gene expression was assessed on the 4th day after transfection. Using this protocol, one small dsRNA (dsRNA-915), which targeted the promoter sequence 915 bases upstream of the transcription start site, was obtained ()). Potent activation of the INTS6 gene by dsRNA-915 was observed in various benign and malignant human cell lines (), which indicated a conservative effect of RNAa in human cells.
Figure 2. A small dsRNA (dsRNA-915) induces INTS6 expression in various human cells. (a) A schematic of the INTS6 promoter showing the transcription start site and the target site of dsRNA-915. (b) CRPC cells (PC3 and DU145), human prostate epithelial cells (RWPE1), human hepatocellular carcinoma cells (HepG2), and normal hepatic cells (L02) were transfected with small RNAs for 96 h at a concentration of 20 nM. INTS6 mRNA was detected by real-time quantitative PCR. GAPDH was used as an internal control. The results were standardized to those of the mock group (transfected with vector alone), and the data are presented as the mean ± SD of three independent experiments. *P < 0.05 compared with the mock group. (c) INTS6 protein was detected by western blot analysis. GAPDH served as a loading control. Increased INTS6 protein expression was observed in dsRNA-915-treated cells
Unique time course and epigenetic modulation were observed for dsRNA-induced INTS6 gene activation
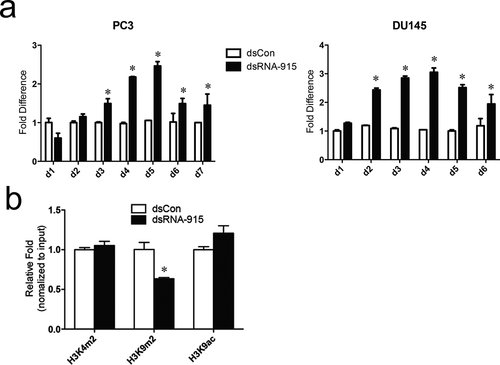
Delayed and prolonged activation of target genes, which is quite different from the kinetics of RNAi or miRNA gene regulation, was observed in RNAa studies[Citation6,Citation8,Citation12,Citation24]. Histone modifications in targeted chromosome regions, such as upregulation of H3K4 methylation, downregulation of H3K9 methylation, and altered H3K9 acetylation, were reported in response to RNAa [Citation6,Citation9,Citation11]. In this study, dose- and time-dependent effects of dsRNA were assessed. At a moderate concentration (20 nM) for transfection, dsRNA-915 activated INTS6 gene transcription in 2 ~ 3 days and maintained this effect for another 5 days ()). The dual-methylation of H3K9 surrounding the dsRNA-915 target site decreased during the period of gene activation ()), which is consistent with previous studies[Citation6,Citation11].
Figure 3. (a) The course of INTS6 activation. PC3 and DU145 cells were transfected with 20 nM dsRNA-915, and INTS6 mRNA was detected every 24 h using real-time quantitative PCR. GAPDH served as an internal control. (b) The effects of dsRNA-915 on histone modifications at the INTS6 promoter in PC3 cells. Using antibodies against H3K4m2, H3K9m2 and H3K9ac, DNA fragments that cross-linked with the specific histone were pulled down and analyzed in ChIP assays. The co-precipitated DNA was amplified by qPCR using a primer set specific for the indicated region (−849/−1022). Input DNA was used as the internal control. The results were standardized to those of the dsControl group, and the data are presented as the mean ± SD of three independent experiments. * P < 0.05 compared with the dsControl group
DsRNA-915 repressed the viability of CRPC cells but not of normal epithelial cells
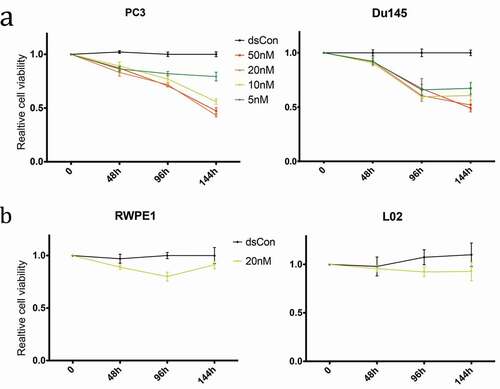
Quiescent cell morphology (round cells with less protrusions) was observed in CRPC cells treated with dsRNA-915 (Figure S1). Then, cell viability was monitored by CCK8 assay. PC3 and DU145 cells treated with different concentrations of dsRNA-915 (5 nM, 10 nM, 20 nM and 50 nM) showed a gradual decrease in viability, particularly in the 20 nM group ()). To assess the effect of dsRNA-915 in benign epithelial cells, the prostate epithelial cell line RWPE1 and the liver epithelial cell line L02 were also transfected with this dsRNA. When treated with 20 nM dsRNA-915, neither benign cell line showed a significant decrease in viability ()), indicating a specific suppressor role for dsRNA-915 in cancer cells.
Figure 4. Time- and dose-dependent inhibition of growth and viability by dsRNA-915 in different cell lines. Cell growth/viability was analyzed using the CCK8 assay. The results were standardized to the mock group, and the data are presented as the mean ± SD (n = 4). (a) Reduced cell growth/viability was observed in dsRNA-915-treated (5, 10, 20 and 50 nM) CRPC cells starting at 48 h. (b) In the benign epithelial cells (RWPE1 and L02), dsRNA-915 (20 nM) moderately inhibited cell growth and viability
DsRNA-915 suppressed clonogenicity and induced cell cycle arrest in CRPC cell lines
Colony formation assays were performed with PC3 and DU145 cells to determine the effect of dsRNA-915 on cell proliferation. Notably, both cell lines showed a significant inhibition of clonogenicity in the dsRNA-915-treated groups ()). Cell cycle was analyzed by flow cytometry, and a significant accumulation of cells in the G1 phase was observed in both cell lines after dsRNA-915 treatment ()). To further evaluate the effect of dsRNA-915 on apoptosis, cells were counted by flow cytometry following Annexin V and PI double staining. Both early and late apoptosis events were rare after dsRNA-915 treatment (Figure S2a). The apoptosis-sensitive protein PARP (poly ADP-ribose polymerase) was also detected by western blot assay. PARP cleavage did not significantly increase in dsRNA-915-treated cells, indicating weak induction of apoptosis in these cells after INTS6 activation (Figure S2b).
Figure 5. (a) Decreased clonogenicity of CRPC cells treated with dsRNA-915. Four hundred cells transfected with dsControl RNA or dsRNA-915 were plated in each well. Two weeks later, the colonies were fixed and stained with crystal violet. Images of the wells are shown, and the data from three independent experiments are presented as the mean ± SD on the right side. (b) Cell cycle arrest caused by dsRNA-915 in CRPC cells. Cells transfected with dsControl RNA or dsRNA-915 were subjected to flow cytometry for cell cycle analysis. Three independent experiments were performed for each group, and the mean percentage of cells in each phase of the cell cycle is presented. The data that reached significance are labeled with asterisks. (c) dsRNA-915 decreased the motility of CRPC cells. PC3 and DU145 cells were transfected with dsControl RNA or dsRNA-915 for 72 h, and cell migration and invasion were assessed by transwell assays. Representative microscopic (200×) migration and invasion images are shown in the upper section. The number of migrating and invading cells was counted, and the results of 3 independent experiments are presented as the mean ± SD in the bottom section (* P < 0.05)
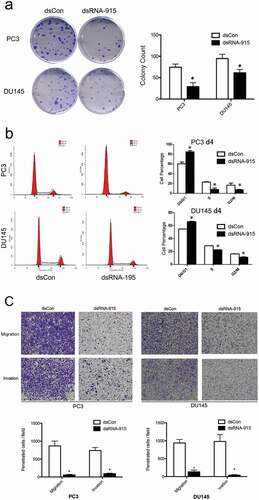
DsRNA-915 decreased the motility of CRPC cells
Tumor metastasis, which is associated with cell invasion and migration, is considered one of the main causes of death among CRPC patients. To evaluate the effect of dsRNA-915 on cell invasion and migration, transwell assays were performed with CRPC cell lines. After a 24-h incubation, a significant decrease in cell migration and invasion was observed in the dsRNA-915-treated group, demonstrating the suppressive effect on motility in these cells ()).
The induction of INTS6 suppressed Wnt signaling in CRPC cell lines
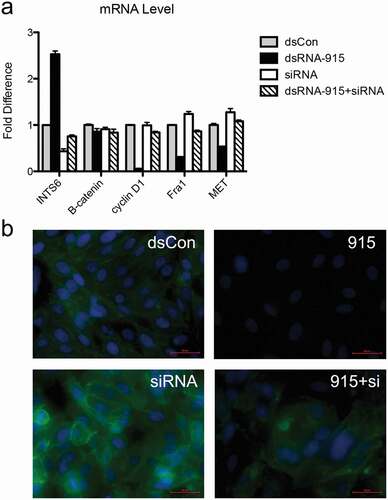
A previous expression profiling study revealed that several genes in the Wnt signaling pathway, such as CXXC finger 4, frizzled homologue 7, transcription factor 7-like 1, and cyclin D1, were regulated in CRPC cells in which INTS6 expression was restored[Citation22]. In our study, β-catenin, which is the key protein in the Wnt signaling pathway, was downregulated post-transcriptionally in dsRNA-915-treated CRPC cells. The downstream targets of β-catenin, such as MET, Fra1, and cyclin D1, were downregulated simultaneously (). Off-target effects can interfere with small RNA target identification. To elucidate whether the suppression of Wnt signaling was an off-target effect or a direct effect of dsRNA-915, this particular small RNA was transfected into PC3 cells in which the INTS6 gene was simultaneously knocked down by siRNA. The downregulation effect of dsRNA-915 treatment on β-catenin signaling was attenuated in INTS6-silenced PC3 cells, suggesting that the regulation of Wnt signaling by dsRNA-915 was mediated by INTS6 expression but not an off-target effect of the small RNA ().
Figure 6. dsRNA-915 suppresses the Wnt signaling pathway in CRPC cells. PC3 and DU145 cells were treated with mock, dsControl RNA, or dsRNA-915 96 h prior to further analysis. (a) The mRNA levels of representative genes in the Wnt signaling pathway were detected using real-time quantitative PCR. The mRNA levels of cyclin D1, Fra1 and MET decreased after dsRNA-915 treatment, whereas those of β-catenin, c-Myc, or VEGF did not decrease. The results were standardized to those of the mock group, and the data are presented as the mean ± SD of three independent experiments. (b) The protein levels of representative genes were detected by western blot assay. Decreased expression of β-catenin, cyclin D1, Fra1 and MET was detected. (c) The immunofluorescent staining of β-catenin in PC3 and DU145 cells is presented. A significant decrease in intracellular β-catenin expression was observed (×200 magnification)
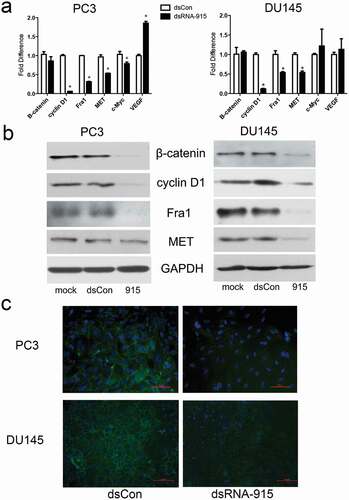
Figure 7. Knockdown of INTS6 reverses the suppressive effect of dsRNA-915 on Wnt/β-catenin signaling in PC3 cells. INTS6 gene expression was knocked down with siRNA targeting the INTS6 mRNA sequence. The ability of dsRNA-915 to suppress Wnt/β-catenin signaling in PC3 cells was assessed with or without siRNA treatment. (a) The siRNA treatment suppressed the up-regulation of INTS6 mRNA in dsRNA-915-treated cells. This siRNA treatment reversed the suppressive effect of dsRNA-915 on Wnt-targeted signals (cyclin D1, Fra1 and MET). The results were standardized to those of the dsControl group, and the data are presented as the mean ± SD of three independent real-time PCR experiments. (b) The β-catenin protein (green fluorescence) was analyzed by immunohistochemistry. The siRNA treatment enhanced β-catenin expression in PC3 cells and partially reversed the suppressive effect of dsRNA-915 on β-catenin protein levels
Deregulation of β-catenin decreased the tumor suppressor effect of dsRNA-915
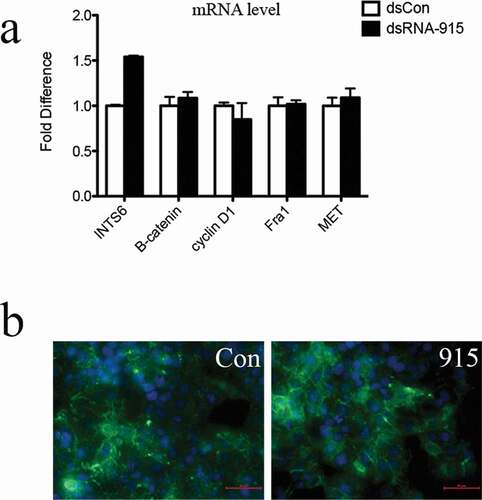
The exact INTS6-regulatory component in Wnt/β-catenin signal remains unknown. To identify the regulatory site, the human hepatocellular carcinoma cell line HepG2, in which mutated β–catenin protein fails to undergo degradation and constitutively activates tumor genes, was used. In this cell line, the aberrant intracellular accumulation of β-catenin protein was observed. Despite the upregulation of INTS6 by dsRNA-915, the consistent expression of Wnt downstream targets (cyclin D1, Fra1 and MET) was detected (). Additionally, the cell cycle distribution and cell mobility were not affected by dsRNA-915 in HepG2 cells (Figure S3). These data suggest that the physiological degradation of β-catenin may be the downstream regulatory site for the INTS6 signal.
Figure 8. Upregulation of INTS6 gene by dsRNA-915 does not inhibit Wnt signaling in HepG2 hepatocellular carcinoma cells. (a) INTS6 mRNA levels increased after dsRNA-915 treatment in HepG2 cells. However, the expression of Wnt-targeted signals (cyclin D1, Fra1 and MET) did not change. The results were standardized to those of the dsControl group, and the data are presented as the mean ± SD of three independent real-time PCR experiments. (b) The β-catenin protein was analyzed using a green fluorescent reagent. The aberrant expression and intracellular localization of β-catenin protein were observed despite dsRNA-915 treatment in HepG2 cells
Discussion
Small RNA-induced gene activation has emerged as a new gene manipulation technique. In the first RNAa study, which was performed in PCa cells, the tumor suppressor genes p21 and E-cadherin were induced by small dsRNAs targeting noncoding regions in gene promoters[Citation6]. Later studies demonstrated that the induction of p21 or E-cadherin expression by small activating RNA leads to the suppression of various tumor cells[Citation13,Citation14,Citation25–Citation27]. In Cas-based transcription activation (the latest targeted gene activation technique), a small RNA is used to guide the activation complex to the promoter region[Citation28,Citation29]. In this study, the defective tumor suppressor gene INTS6 was induced in CRPC cells by RNAa and tumor suppression was observed in treated cells. These data suggest that RNAa is a novel strategy for targeted cancer therapy. Downregulated tumor suppressor genes in cancer should be carefully selected as potential therapeutic targets of RNAa. For RNA activation, intact and proper genetic information of the targeted gene should be maintained in the genome, or at least in one of the alleles because saRNA occurs at the transcriptional level. The frequent downregulation of INTS6 in PCa is most likely due to the double-hit of LOH and abnormal epigenetic modulation[Citation23] rather than mutation[Citation30,Citation31], which fulfills the criteria of an RNAa target.
The optimal design strategy for small activating RNAs remains unclear. In most studies, small activating RNAs were designed according to promoter sequences, but gene expression induced by small RNAs complementary to the 3ʹ terminus was also reported[Citation32]. The algorithm developed for the design of siRNAs was applied in the design of saRNA[Citation6,Citation11], which suggests a similar mechanism of action of these two types of small RNAs in gene regulation. Homology to other known human sequences should be avoided in the saRNA design because off-target effects can occur at homologous sites. Because noncoding transcripts overlapping the promoter region may enable recognition by and localization of small RNAs[Citation16,Citation24], saRNAs complementary to the overlapping region may be more potent in gene induction. In this study, the promoter region of INTS6 (200 ~ 1000 bp upstream of the transcription start site), where a noncoding transcript INTS6-AS1 overlaps[Citation33], was carefully selected for the saRNA design. The conservation and potency of dsRNA-915-induced INTS6 gene activation in different cell lines strongly support these recent design principles. More efficient saRNA design criteria will be developed based on accumulating RNAa knowledge.
The mechanism of small RNA-induced activation continues to be studied. The identification of RNAa was once questioned, as small RNAs may upregulate supposed target genes indirectly via off-target effects[Citation34]. However, RNAa is always coupled to epigenetic modulation around the targeted chromatin site [Citation6,Citation7,Citation10,Citation11,Citation27,Citation29,Citation32], which cannot be fully explained by the off-target theory. In addition, gene induction by saRNA appears 24 ~ 48 h later than gene suppression by siRNA or miRNA[Citation12]. Moreover, gene upregulation by saRNA is sustained for as long as 1 ~ 2 weeks[Citation6,Citation12,Citation32], greatly surpassing the time frame for small RNA metabolism. These features, which were also observed in this study, suggest a unique mechanism of RNAa action.
Research on Argonaute (AGO) proteins and noncoding RNAs has promoted the study of RNAa. AGO proteins are the catalytic components of the RNA-induced silencing complex (RISC), the protein complex responsible for RNAi)[Citation35]. Although different AGO proteins are utilized in RNAi, only AGO2 is essential in the process of RNAa [Citation6,Citation12,Citation36]. The accumulation of AGO2 protein in the promoter region has been observed with RNAa[Citation6,Citation10,Citation12]. Interestingly, binding between AGO2 and noncoding transcripts overlapping the promoter region has been reported in RNAa studies. Instead of splicing these transcripts, the saRNA-AGO2 complex modulates the heterochromatin in the targeted DNA region[Citation24]. A reasonable hypothesis is that AGO2 participates in the maturation of saRNA by a similar process as that which occurs for siRNA, thus forming an RNA-AGO complex. Then, the complex recognizes the promoter region with or without the help of noncoding RNA transcripts and elicits activating changes in the promoter. Further studies should focus on the detailed mechanism by which saRNAs recognize and modify targeted DNA.
The Wnt signaling pathway is crucial in a variety of biological processes. The intracellular accumulation and nuclear translocation of β-catenin is the key signal transduced in the canonical Wnt signaling pathway[Citation37]. The aberrant localization of β-catenin is frequently detected in PCa, particularly in advanced, metastatic or hormone-refractory lesions[Citation38–Citation41]. The intracellular overexpression of β-catenin may facilitate the nuclear transduction of androgen receptor (AR) signaling[Citation40,Citation42,Citation43], promote stem cell self-renewal[Citation44], drive cell invasion and migration[Citation45,Citation46], and ultimately cause castration-resistant or metastatic disease. The link between INTS6 expression and Wnt signaling was first reported by Filleur et al[Citation22]. In their study, exogenous re-expression of INTS6 in CRPC cells suppressed cell growth. During this process, Wnt signals, such as CXXC4, FZD7 and TCF7L1, were upregulated, whereas cyclin D1 and TCF7 were clearly downregulated. In our study, consistent results, such as cell cycle arrest and suppression of Wnt signaling, were observed in response to INTS6 induction. Moreover, we revealed that upregulation of INTS6 decreased the motility of CRPC cells and that downregulation of β-catenin participated in the processes by which INTS6 exerted its tumor suppressor function. As the genetic mutation that interrupts β-catenin degradation is relatively rare in prostate cancer[Citation47,Citation48], the physiological downregulation of β-catenin by INTS6 may be a potential therapeutic strategy in CRPC treatment. The underlying mechanisms by which INTS6 regulates β-catenin and the translation of tumor-suppressor saRNAs into clinical applications deserve further study.
Materials and methods
Human prostate cancer tissue arrays
Prostate cancer tissue arrays (OD-CT-UrPrt03-001) were obtained from Shanghai Outdo Biotech Company. The array contained 28 cases of primary prostate adenocarcinoma. Malignant prostate tissue and adjacent benign tissue were embedded in pairs in each case. All experiments involving human tissues were conducted in accordance with protocols approved by the Medical Ethics Committee of Zhejiang University. The tissue was pathologically reviewed by a pathologist.
Immunohistochemistry
Immunostaining was performed in a two-step manner using a DAKO EnVision Kit (Dako Co., GK500705) according to the manufacturer’s instructions. Microwave heating in sodium citrate solution (pH 6.0) was performed for epitope retrieval. The INTS6 primary antibody was purchased from Santa Cruz Biotechnology (sc-376,524). The images were obtained under a microscope (Olympus, Tokyo, Japan) with appropriate magnification, and brown DAB staining was considered positive. In the assessment, the proportion of positive cells (less than 5% positive nuclei, 5 ~ 20% positive nuclei, and over 20% positive nuclei) and the average staining intensity (blue, yellow and brown nuclei) were graded as 0, 1, or 2, with 0 representing the lowest score and 2 representing the highest score. The proportion and intensity results were multiplied and evaluated as follows: 4, positive staining (+); 1 to 2, decreased staining (±); and 0, negative staining (-).
Cell lines, cell culture, and small RNA transfection
The human castration-resistant prostate cancer cell lines PC3 and DU145, the human hepatocellular carcinoma cell line HepG2, the human prostate epithelial cell line RWPE1, and the human hepatic cell L02 were obtained from the Shanghai Institute of Cell Biology (Shanghai, China). Cells were cultured in RPMI 1640 medium (PC3, DU145, RWPE1, and L02) or DMEM (HepG2) supplemented with 10% (v/v) FBS in a 5% CO2 atmosphere at 37°C.
Small RNA was transfected using Lipofectamine 2000 (Thermo Fisher, 11,668,019) according to the manufacturer’s protocol. Briefly, small RNA was co-incubated with liposomes in serum-free medium prior to transfection. Then, the pre-assembled RNA-liposome complexes were added into the cell culture medium and delivered the small RNA into the cells. Double-strand small activating RNA (dsRNA-915), nonsense double-strand control RNA and vector control (Lipofectamine 2000 alone) were utilized in the saRNA, dsControl and mock groups, respectively. All small RNAs were chemically synthesized by GenePharma Co. (Shanghai, China), and the sequences are provided in the Supporting Information (Table S2).
RNA extraction and quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Exiqon, 300,110) and reverse transcribed into cDNA using a PrimeScript RT Reagent Kit (Takara, RR037A). The resulting cDNA was detected with an ABI 7500 real-time PCR System (Applied Biosystems, Carlsbad, USA) using SYBR Green (Clontech, 639,676). GAPDH mRNA served as the internal control. The qPCR primers were obtained from Sango Biotech (Shanghai, China), and the sequences are listed in the Supporting Information (Table S2).
Western blot assay
Briefly, whole cell lysates were denatured, resolved by electrophoresis in 10–15% Tris–glycine polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked and then incubated with primary antibodies against INTS6 (Santa Cruz Co., sc-376,524), PARP (Epitomics, S0164), β-catenin (Epitomics, 1247–1), Fra1 (Epitomics, 5136–1), MET (Epitomics, 1996–1), or cyclin D1 (Epitomics, 2261–1) at dilutions specified by the manufacturer. Then, the membranes were incubated with the corresponding HRP (horseradish peroxidase)-conjugated secondary antibody, and the bound secondary antibody was detected using an enhanced chemiluminescence system (Pierce Biotechnology, Inc., Rockford, IL, USA).
Immunocytofluorescence
Cells were fixed in absolute methanol for 20 min, blocked in 5% BSA, and incubated with a primary antibody against β-catenin (Epitomics, 1247–1). A FITC-labeled secondary antibody (Abcam, ab6717) was used for the immunofluorescence assay. After immunolabeling, the cells were washed, stained with DAPI (Sigma, D9542), and viewed using a fluorescence microscope (Nikon).
Chip assay
ChIP assays were performed using an EZ-Magna ChIP Assay Kit (MerckMillipore, 17–371) according to the manufacturer’s instructions. Briefly, cells (2 × 106) were plated for 48 h and treated with control RNA or dsRNA-915 for 48 h. Formaldehyde treatment was used to cross-link the proteins to the DNA. Cells were then lyses and sonication was performed to shear the chromatin to 200 ~ 1000 bp size. Antibodies that recognize H3K9m2, H3K4m2 and H3K9ac (Millipore, 17–648, 17–677 and 17–658) were used to immunoprecipitate the chromatin debris to detect specific changes in histone methylation and acetylation patterns. Immunoprecipitated DNA was reverse cross-linked, purified, and analyzed by qPCR. PCR primers used for ChIP analysis are described in the Supporting Information (Table S2).
Cell growth/viability assay
Cells were seeded in 96-well plates at a density of 4 × 103/well. After an overnight incubation, the cells were treated with RNA duplexes (5–50 nM) for 0–144 h. Cell counting solution (CCK-8, Dojindo Laboratories, CK04-05) was added to each well at the endpoint. After an additional 2-h incubation, the absorbance of the solution was measured spectrophotometrically at 450 nm with an MRX II absorbance reader (Dynex Technologies, Chantilly, VA, USA). The data were standardized to that of the mock group.
Colony formation assay
The cells were harvested 24 h after transfection with RNA duplexes (20 nM). Four hundred transfected cells were seeded in a fresh six-well plate and cultured undisturbed for 2 weeks, during which time the surviving cells spawned a colony of proliferating cells. The cell colonies were stained with 0.1% crystal violet, and colonies with a diameter greater than 2 mm were counted.
Cell cycle and apoptosis assays by flow cytometry
Cells were harvested 96 h after transfection. For cell cycle assays, the cells were fixed in 75% ethanol, stained with DNA Prep Stain (Beckman Coulter, 6,607,055), and counted using a BD LSRII Flow Cytometry System with FACSDiva software (BD Biosciences, Franklin Lakes, NJ, USA). The raw data were analyzed with ModFit LT 3.2 software (Verity Software House, Topsham, ME, USA). For cell apoptosis assays, Annexin V and PI double staining was performed using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, 556,547) according to the manufacturer’s protocol. Then, the BD LSRII Flow Cytometer System was used for the apoptosis analysis.
Transwell assay
The cells were harvested 72 h after transfection, and 8 × 104 cells were added to the top chamber of a well containing an uncoated (for migration assays) or a Matrigel-coated (for invasion assays, BD Matrigel, 356,234) PET membrane (24-well insert, 8-mm pore size, BD Falcon, 353,095). Medium supplemented with 20% fetal bovine serum was added to the lower chamber of each well to act as a chemo-attractant. The cells were incubated for 24 h, and those that reached the lower surface of the membrane were fixed in 100% methanol and stained with 0.1% crystal violet. These number of invading cells in five random fields was counted under a microscope, and the average number of cells per field was recorded.
Statistics
The statistical analyses were performed using SPSS 20.0 statistical software (SPSS, Chicago, IL, USA). Student’s t-test followed by Dunnett’s multiple comparison test was used to compare quantitative data. The Wilcoxon test (for paired data) and Mann-Whitney (for grouped data) nonparametric test were used to compare categorical data. Values of P < 0.05 were considered significant, and these significant differences are indicated by asterisks in the Figures.
Supplemental Material
Download Zip (11.3 MB)Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81372773) and the Natural Science Foundation of Zhejiang Province (Grant No. LQ16H160011 & No. LY16H160015). We also thank M.D. Xiao-Dong Ten, from the Pathology Department of the First Affiliated Hospital of Zhejiang University, for his generous help on pathological assessment.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
Additional information
Funding
Related Research Data
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015 Mar;65(2):87–108. PMID: 25651787.
- Pagliarulo V, Bracarda S, Eisenberger MA, et al. Contemporary role of androgen deprivation therapy for prostate cancer. Eur Urol. 2012 Jan;61(1):11–25. PMID: 21871711.
- Cookson MS, Roth BJ, Dahm P, et al. Castration-Resistant prostate cancer: AUA guideline. J Urol. 2013 May 9. PMID: 23665272. DOI:https://doi.org/10.1016/j.juro.2013.05.005
- Mottet N, Bellmunt J, Bolla M, et al. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Actas Urol Esp. 2011 Nov-Dec;35(10):565–579. PMID: 21757258.
- Clarke NW. Landmarks in non-hormonal pharmacological therapies for castration-resistant prostate cancer. BJU Int. 2012 Oct;110(Suppl 1):14–22. PMID: 23046036.
- Li LC, Okino ST, Zhao H, et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A. 2006 Nov 14;103(46):17337–17342. PMID: 17085592.
- Janowski BA, Younger ST, Hardy DB, et al. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007 Mar 3;3:166–173.
- Huang V, Qin Y, Wang J, et al. RNAa is conserved in mammalian cells. PloS ONE. 2010;5(1):e8848.
- Place RF, Li LC, Pookot D, et al. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008 Feb 5;105(5):1608–1613. PMID: 18227514.
- Turunen MP, Lehtola T, Heinonen SE, et al. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy. Circ Res. 2009 Sep 11;105(6):604–609. PMID: 19696410.
- Yang K, Shen J, Xie YQ, et al. Promoter-targeted double-stranded small RNAs activate PAWR gene expression in human cancer cells. Int J Biochem Cell Biol. 2013 Jul;45(7):1338–1346. PMID: 23583662.
- Place RF, Noonan EJ, Foldes-Papp Z, et al. Defining features and exploring chemical modifications to manipulate RNAa activity. Curr Pharm Biotechnol. 2010 Aug;11(5):518–526. BSP/CPB/E-Pub/0094-11-6aaa [pii]. PMID: 20662764.
- Wei J, Zhao J, Long M, et al. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer. 2010;10:632. PMID: 21087528.
- Junxia W, Ping G, Yuan H, et al. Double strand RNA-guided endogeneous E-cadherin up-regulation induces the apoptosis and inhibits proliferation of breast carcinoma cells in vitro and in vivo. Cancer Science. 2010 Aug;101(8):1790–1796. PMID: 20518789.
- Kang MR, Yang G, Place RF, et al. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res. 2012 Oct 1;72(19):5069–5079. PMID: 22869584.
- Matsui M, Sakurai F, Elbashir S, et al. Activation of LDL receptor expression by small RNAs complementary to a noncoding transcript that overlaps the LDLR promoter. Chem Biol. 2010 Dec 22;17(12):1344–1355. PMID: 21168770.
- Wieland I, Arden KC, Michels D, et al. Isolation of DICE1: a gene frequently affected by LOH and downregulated in lung carcinomas. Oncogene. 1999 Aug 12;18(32):4530–4537. PMID: 10467397.
- Yin Z, Spitz MR, Babaian RJ, et al. Limiting the location of a putative human prostate cancer tumor suppressor gene at chromosome 13q14.3. Oncogene. 1999 Dec 9;18(52):7576–7583. PMID: 10602517.
- Li WJ, Hu N, Su H, et al. Allelic loss on chromosome 13q14 and mutation in deleted in cancer 1 gene in esophageal squamous cell carcinoma. Oncogene. 2003 Jan 16;22(2):314–318. PMID: 12527901.
- Melamed J, Einhorn JM, Ittmann MM. Allelic loss on chromosome 13q in human prostate carcinoma. Clin Cancer Res. 1997 Oct 3;10:1867–1872. PMID: 9815575.
- Cooney KA, Wetzel JC, Merajver SD, et al. Distinct regions of allelic loss on 13q in prostate cancer. Cancer Res. 1996 Mar 1;56(5):1142–1145. PMID: 8640774.
- Filleur S, Hirsch J, Wille A, et al. INTS6/DICE1 inhibits growth of human androgen-independent prostate cancer cells by altering the cell cycle profile and Wnt signaling. Cancer Cell Int. 2009;9:28. PMID: 19906297.
- Ropke A, Buhtz P, Bohm M, et al. Promoter CpG hypermethylation and downregulation of DICE1 expression in prostate cancer. Oncogene. 2005 Oct 6;24(44):6667–6675. PMID: 16007164.
- Schwartz JC, Younger ST, Nguyen NB, et al. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol. 2008 Aug;15(8):842–848. PMID: 18604220.
- Chen Z, Place RF, Jia ZJ, et al. Antitumor effect of dsRNA-induced p21(WAF1/CIP1) gene activation in human bladder cancer cells. Mol Cancer Ther. 2008 Mar 7;(3):698–703. PMID: 18347154.
- Mao Q, Zheng X, Yang K, et al. Suppression of migration and invasion of PC3 prostate cancer cell line via activating E-cadherin expression by small activating RNA. Cancer Invest. 2010 Dec;28(10):1013–1018. PMID: 20690797.
- Yang K, Zheng XY, Qin J, et al. Up-regulation of p21WAF1/Cip1 by saRNA induces G1-phase arrest and apoptosis in T24 human bladder cancer cells. Cancer Lett. 2008 Jul 8;265(2):206–214. PMID: 18358601.
- Cheng AW, Wang H, Yang H, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013 Oct;23(10):1163–1171. PMID: 23979020.
- Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014 Mar 14;343(6176):1247997. PMID: 24505130.
- Hernandez M, Papadopoulos N, Almeida TA. Absence of mutations in DICE1/DDX26 gene in human cancer cell lines with frequent 13q14 deletions. Cancer Genet Cytogenet. 2005 Nov;163(1):91–92. PMID: 16271964.
- Bohm M, Maier C, Kufer R, et al. Genetic variants of DICE1/INTS6 in German prostate cancer families with linkage to 13q14. Urol Int. 2015;954:386–389. PMID: 25660097.
- Yue X, Schwartz JC, Chu Y, et al. Transcriptional regulation by small RNAs at sequences downstream from 3ʹ gene termini. Nat Chem Biol. 2010 Aug 6;(8):621–629. PMID: 20581822.
- Peng H, Ishida M, Li L, et al. Pseudogene INTS6P1 regulates its cognate gene INTS6 through competitive binding of miR-17-5p in hepatocellular carcinoma. Oncotarget. 2015 Mar 20;6(8):5666–5677. PMID: 25686840.
- Weinberg MS, Barichievy S, Schaffer L, et al. An RNA targeted to the HIV-1 LTR promoter modulates indiscriminate off-target gene activation. Nucleic Acids Res. 2007;35(21):7303–7312. PMID: 17959645.
- Cenik ES, Zamore PD. Argonaute proteins. Current Biology: CB. 2011 Jun 21;21(12):R446–9. PMID: 21683893.
- Morris KV, Santoso S, Turner AM, et al. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008 Nov 4;(11):e1000258. PMID: 19008947.
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009 Jul;17(1):9–26. PMID: 19619488.
- Morita N, Uemura H, Tsumatani K, et al. E-cadherin and alpha-, beta- and gamma-catenin expression in prostate cancers: correlation with tumour invasion. Br J Cancer. 1999 Apr;79(11–12):1879–1883. PMID: 10206308.
- Chen G, Shukeir N, Potti A, et al. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer. 2004 Sep 15;101(6):1345–1356. PMID: 15316903.
- Schweizer L, Rizzo CA, Spires TE, et al. The androgen receptor can signal through Wnt/beta-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol. 2008;9:4. PMID: 18218096.
- Wan X, Liu J, Lu JF, et al. Activation of beta-catenin signaling in androgen receptor-negative prostate cancer cells. Clin Cancer Res. 2012 Feb 1;18(3):726–736. PMID: 22298898.
- Mitani T, Harada N, Nakano Y, et al. Coordinated action of hypoxia-inducible factor-1alpha and beta-catenin in androgen receptor signaling. J Biol Chem. 2012 Sep 28;287(40):33594–33606. PMID: 22865883.
- Wang G, Wang J, Sadar MD. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008 Dec 1;68(23):9918–9927. PMID: 19047173.
- Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009 Jun;19(6):683–697. PMID: 19365403.
- Francis JC, Thomsen MK, Taketo MM, et al. beta-catenin is required for prostate development and cooperates with Pten loss to drive invasive carcinoma. PLoS Genet. 2013;9(1):e1003180. PMID: 23300485.
- Dulinska-Litewka J, McCubrey JA, Laidler P. Increased Akt signaling resulting from the loss of androgen responsiveness in prostate cancer. Curr Med Chem. 2013;20(1): 144–157. PMID: 23033951.
- Chesire DR, Ewing CM, Sauvageot J, et al. Detection and analysis of beta-catenin mutations in prostate cancer. The Prostate. 2000 Dec 1;45(4):323–334. PMID: 11102958.
- Voeller HJ, Truica CI, Gelmann EP. Beta-catenin mutations in human prostate cancer. Cancer Res. 1998 Jun 15;58(12):2520–2523. PMID: 9635571.